Core Pathways of Aging
Most overviews of aging suffer from multiple problems:
They dump a bunch of findings with no high-level picture.
Many of the claims they make are outdated, purely theoretical, and sometimes even outright disproven by existing work.
They are usually written by working academics, who are shy about telling us when their peers’ work is completely wrong.
They are shy about making strong claims, since this would also implicitly mean denying some claims of the authors’ peers.
This post is a high-level brain-dump of my current best models of the core pathways of aging, as I currently understand them. I have no particular reason to avoid calling out claims I think are wrong/irrelevant, and I’m going to present high-level models without pages and pages of disclaimers and discussions about results which maybe disagree with them (but are probably just wrong/irrelevant).
Epistemic status: I would be surprised if none of it turned out to be wrong, but there are multiple lines of evidence supporting most claims. It is not highly polished, and references are included only when I have them readily on hand. My ideal version of this piece would have more detailed references, more double-checking behind the claims, and more direct presentation of the data which backs up each claim. Unfortunately, that would take enough time and effort that I’m unlikely to actually get to it soon. So… here’s what I could produce in a reasonable amount of time. Hopefully it will be wrong/unhelpful in ways orthogonal to how most overviews are wrong/unhelpful.
Foundations
First, let’s recap a couple foundational principles. I’ll go through these pretty quickly; see the linked posts for more info.
Homeostasis and “Root Causes” in Aging: the vast majority of proteins, cells, etc, in the human body turn over on a timescale from days to months. At any given time, their level (e.g. protein concentration, cell count, etc) is in equilibrium on the turnover timescale—i.e. the rate of creation approximately equals the rate of removal. For any X with turnover much faster than aging (i.e. decades), if we see the level of X increase/decrease on the timescale of a human lifetime, then that is not due to permanent “accumulation of X” or “depletion of X”; it is due to increase/decrease in the rate of creation/removal of X. For instance:
DNA damage is typically repaired on a timescale of hours or faster, depending on the type. If DNA damage levels increase with age, that is due to an increase in rate of damage or decrease in rate of repair, not permanent accumulation.
Typical senescent cells turn over on a timescale of days to weeks. If the number of senescent cells increases with age, that is due to an increase in rate of senescent cell production or decrease in rate of removal, not permanent accumulation.
Elastin is believed to not turn over at all in humans. So if we see elastin deposits increasing with age (e.g. in wrinkles), then that could be permanent accumulation.
Furthermore: suppose we have a positive feedback cycle. Increasing A decreases the rate of production of B, so B decreases. But decreasing B decreases the rate of removal of A, so A increases. If both A and B individually turn over on a timescale of hours or faster then this feedback loop as a whole will also typically operate on a timescale of hours or faster—i.e. count/concentration of A will explode upward on roughly that timescale. More generally, a feedback loop will usually operate on the timescale of its slowest component, exactly like the rate-limiting step of a chemical reaction.
Main upshot of all this: since aging involves changes on a timescale of decades, there must be some component which is out-of-equilibrium on a timescale of decades or longer (i.e. does not turn over significantly across a full human lifespan). These are the components which we’ll call “root causes”. Everything else which changes with age, changes only in response to the root causes. Reset the root causes to young-organism levels, and everything else will equilibrate to young-organism levels in response. Furthermore, a reset of the root causes only needs to happen once every few decades for humans—it fully resets the human to a youthful state, so ongoing treatment is not needed on short timescales.
The Lens, Progerias and Polycausality: The lens of the human eye consists of fiber deposits which do not turn over significantly over the course of a human lifetime. New fiber layers are added over time, so the lens grows from around 3.5mm in infancy to 5.5mm in old age. The main clinical result is the near-universal need for reading glasses in old age.
This is a well-understood root cause of one symptom of old age. Furthermore, it is very likely independent of most other age related diseases—lens thickening is unlikely to cause cancer or heart disease, for instance. So, it’s an existence proof: there is more than one root cause of aging.
That said, there’s a fair bit of evidence that most symptoms of aging—including the major age-related diseases—share a common root cause, or at least a common core pathway. Some kinds of evidence of this:
Most symptoms/diseases of aging are correlated—someone who has one early is likely to have others. Conceptually, if you do a factor analysis on aging symptoms, there’s one big factor for a bunch of diseases, even after controlling for the number of years one has lived. (“Aging clock” is a relevant piece of jargon here.)
At the cellular level, a lot of diseases of aging “look similar”, and involve similar pieces. There’s a decrease in cell count, increase in damaged proteins/DNA/fats, and inflammation. We see roughly this pattern in Alzheimers, atherosclerosis, muscle loss, and many others.
Certain simple interventions reliably produce many diseases of aging—for instance, progerias are single mutations which produce a whole “early aging” phenotype
Conversely, certain simple interventions reliably delay many diseases of aging—e.g. calorie restricted diets.
… so these all point to shared underlying causes. This post will be about the “core pathways” most likely involved.
Major Diseases
These subsections will talk about various specific age-related diseases, mainly highlighting how they connect to the cellular processes we’ll talk about later. Two main themes to watch: reactive oxygen species and senescent cells.
Reactive oxygen species (ROS) aka free radicals are produced in greater numbers in old age. These are short-lived, highly reactive molecules. They react with all sorts of things, oxidatively “damaging” whatever they hit, including proteins, fats, and DNA.
Senescent cells are cells which have partially shut down in a programmatic way, triggered by some sort of “stress” on the cell (e.g. DNA damage, exposure to harsh chemicals or radiation, etc). They pump out inflammatory signals (called the senescence-associated secretory phenotype, or SASP). Eventually, they’re removed by the immune system.
In later sections, we’ll see that these two are tightly coupled. For now, we’ll talk about how they seem to underlie a variety of age-related diseases.
Atherosclerosis
If you dissect young and old mammals, one of the most obvious internal differences is in the blood vessels:
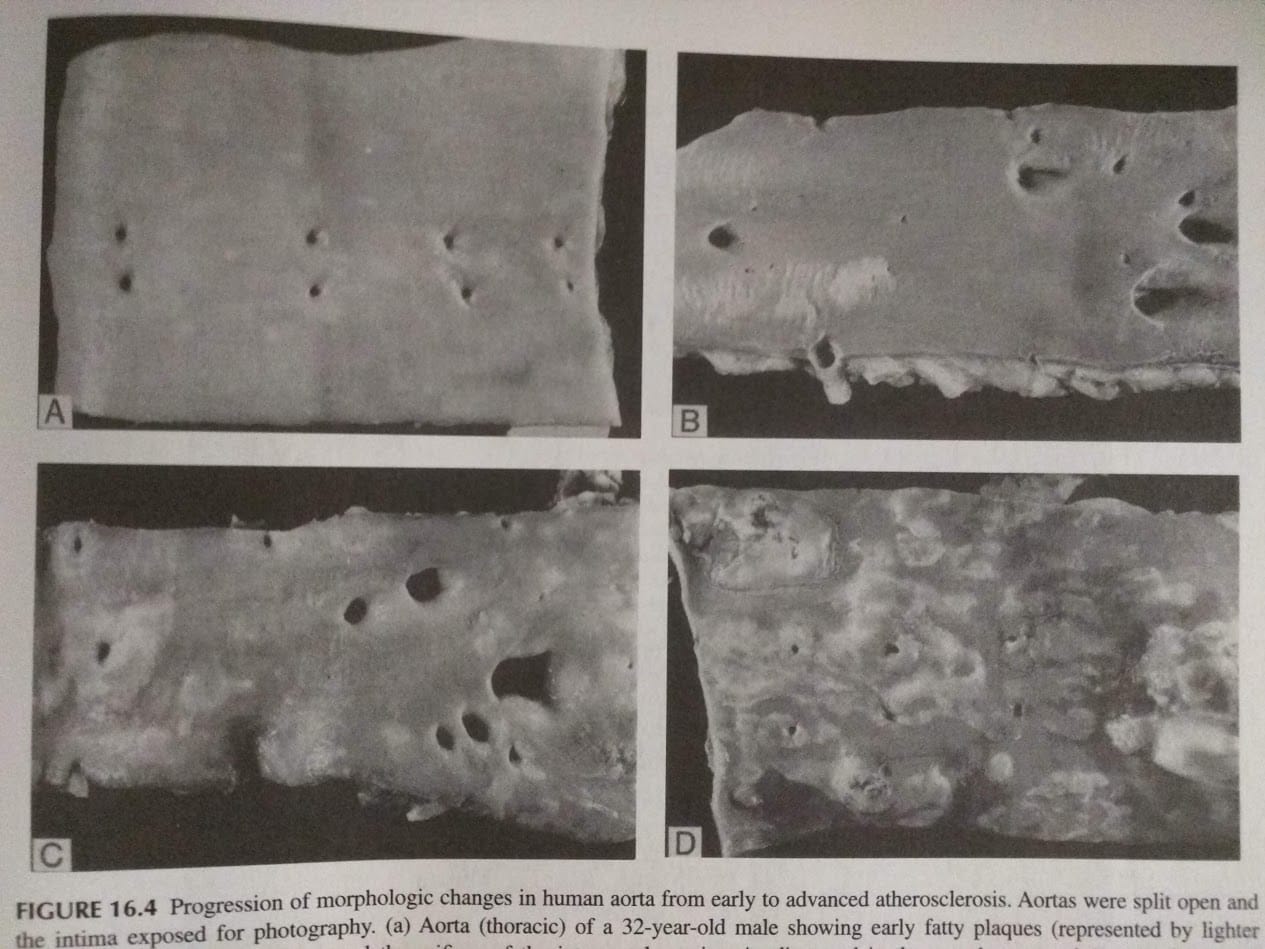
Mammals of any age have “fatty streaks” along the walls of the vasculature, which are exactly what they sound like. (These are the slightly lighter patches in the pictures above—more obvious in the older aortas, but faintly visible in the young pair as well.) In older mammals, the fatty streaks tend to be larger, until in old age they necrotize (aka die) in the middle and turn into thick “atherosclerotic plaques” (the dark patches in the lower right picture above). These can block blood circulation, and sometimes a chunk of the plaque can break off and block circulation in smaller vessels; either of these can cause e.g. heart attack or stroke.
At any age, a lower-fat diet is associated with smaller fatty streaks and lower chance of atherosclerosis, though the streaks universally grow with age holding diet constant.
The big breakthrough in atherosclerosis came in the 80’s/90’s. It was known that a certain type of cell (called a macrophage) would hoover up fat from the bloodstream, then adhere to the cell wall when full, forming the fatty streaks. The missing puzzle piece was that the macrophages don’t just hoover up any random fats (there’s rather a lot of fat in the bloodstream and not that many macrophages; it would be like sweeping sand off the beach). They specifically hoover up partially oxidized fats. Steinberg has a great review of various experiments feeding into this discovery.
The upshot: streaks and plaques grow in old age primarily because the concentration of (partially) oxidized fats in the bloodstream increases in old age. These probably don’t have a very slow turnover time, so either the rate of (partial) oxidation of fats increases in old age, or the rate of removal decreases. Which is it, and what causes it?
The main candidate here is ROS: increasing ROS levels in old age increase the rate of oxidative damage to fats. (One interesting question: how does the relative increase in oxidative damage to fats compare to DNA/proteins? Do the numbers actually line up for these to share a source? I haven’t seen a solid Fermi estimate on this, I’d be interested to see one.)
Vascular Stiffening
Even aside from atherosclerosis, the walls of blood vessels change in old age. In particular they become stiffer.
In normal operation, the heart pumps out blood in discrete chunks—each heartbeat pumps out some amount of blood into the arteries. The arteries expand a bit to accumulate this blood, much like a balloon. And, like an inflated balloon, the arteries are at slightly higher pressure from this extra blood, until it’s pushed out through the capillaries and the cycle starts again with the next heartbeat. It’s sort of like a capacitor: the heart sends out blood at high pressure in sudden waves, and the arteries expand and contract to store the extra blood and smooth out the pressure variance.
If the walls become stiffer, then the arteries are less able to smooth out this pressure variance. From the heart’s perspective, it’s trying to pump blood into a hard container, which works about as poorly as you’d expect; this is one of the major causes of heart failure (alongside atherosclerosis). The vessels also become more likely to burst (i.e. aneurysm).
What causes the loss of elasticity?
The main answer seems to be oxidative damage to proteins in the wall of the blood vessels. The key experimental evidence backing this up is the effect of aminoguanidine (AG), a powerful scavenger of certain ROS: AG administration reverses age-related stiffening of the arteries. This is only temporary—i.e. the arteries go back to their stiff state shortly after AG administration stops—so it isn’t a root cause, but it is a link in the causal chain. Once again, oxidative damage likely caused by increased ROS seems to be the key causal intermediate.
(Please don’t go running out to take aminoguanidine. The side effects are serious, and the benefits are short-term—you’ll go right back to where you would have been as soon as you stop taking it.)
An interesting side note: many sources claim that a shift in collagen:elastin ratio is involved in stiffening of the vasculature. This seems to be based on a purely theoretical paper from decades ago, and the actual data doesn’t back it up, yet review articles and textbooks continue to mention it.
Alzheimer’s
The first and most important thing to know about Alzheimer’s (aka dementia, aka old folks losing their memory) is that it is not caused by accumulation of amyloid beta.
Decades ago, people noticed that if you look at the brains of old people with dementia, they usually have lots of plaques, and these plaques are made of a particular protein fragment called amyloid beta. Therefore clearly amyloid beta causes dementia. Pretty soon people were using amyloid beta plaques to diagnose dementia, which made it really easy to show that the plaques cause dementia: when the plaques are how we diagnose “dementia”, then by golly removing the plaques makes the “dementia” (as diagnosed by plaques) go away.
As far as I can tell, there has never at any point in time been compelling evidence that amyloid beta plaques cause age-related memory problems. Conversely, I have seen at least a few studies suggesting the plaques are not causal.
Meanwhile, according to wikipedia, 244 Alzheimer’s drugs were tested in clinical trials from 2002-2012, mostly targeting the amyloid plaques. Of those, only 1 drug made it through. Bottom line: they don’t work.
So what does cause Alzheimer’s? I don’t know; it’s not a disease I’ve studied in depth. I know plenty of studies find the usual culprits involved—inflammation, damaged proteins, etc.
I do know of one particularly interesting cluster of studies which found that the brain “opens up” during sleep, increasing flow of cerebral fluid to clear out whatever junk accumulated during the day—including amyloid beta. The flow takes place in “paravascular” spaces, i.e. spaces around the blood vessels, which widen during sleep. Given the age-related changes in blood vessels (e.g. thickening & stiffening of walls), it would make sense for this paravascular space to not open up as much in older people, and indeed that seems to be the case. Whether this has anything to do with dementia, I don’t know, but it is my current best guess for the main cause of the amyloid plaques. If true, this would mean that the plaques share their main root causes with changes in the vasculature.
Sarcopenia
Sarcopenia is age-related loss of muscle mass—i.e. old people becoming physically weak. Overviews often say it can be (at least partially) “reversed” via exercise, which seems to be fairly obvious bullshit aimed at making people exercise more. Bottom line: for any fixed amount of exercise, muscular strength will fall with age. (Obviously more exercise can still increase muscle strength at any age.)
Many sources will claim that lots of research has shown sarcopenia to be caused by loss of muscle innervation—i.e. problems with the neuromuscular junctions (NMJs). As with amyloids and Alzheimers, as far as I can tell there was never at any point in time any compelling evidence that NMJ changes are actually causal for muscle loss, and my current best guess is that they are not causal. However, research on age-related changes in NMJ structure did produce very shiny images, which I think is probably the main reason they received so much attention in the first place.
So what does cause sarcopenia? I don’t yet have a full picture here, but I can sketch out my current best guess.
Muscle cells are not normal cells; they’re mega-cells, a hundred times longer than a normal cell, with hundreds of nuclei all within the same cell. They need so many nuclei because molecules which would diffuse quickly from one end to the other of a normal cell would take far too long to diffuse across a muscle cell (diffusion time increases faster-than-linearly with distance); so, things need to be produced locally.
This setup also makes it difficult for an entire muscle cell to turn over. Instead, the nuclei turn over (along with all the usual protein/membrane/etc turnover mechanisms); they’re regularly replaced from “satellite cells”, a type of stem cell nestled in next to the muscle whose job is basically to crank out new nuclei from time to time.
In sarcopenia, one cross-section of the long muscle cell will fail first—a “ragged red” section—and then failure gradually spreads along the length. The failing section involves the usual culprits: ROS, mitochondrial deficiency (related to ROS; more on that later), inflammation signals, etc.
My best guess is that this is basically just how cellular senescence manifests in a muscle cell. One particular satellite cell is “close to senescence”, and produces nuclei which rapidly “senesce”. But since muscle cells are really a bunch of relatively-isolated components along their length (due to long diffusion time), this only results in one section of the cell failing. Eventually, the “senescence” spreads to adjacent sections via the usual mechanisms of senescence-induced-senescence (more on that later). Key characteristics of “ragged red” sections of the muscle cell—like high ROS, mitochondrial deficiency, or inflammation—are the usual characteristics of senescent cells, and sarcopenia probably shares its root cause(s) with cellular senescence more generally.
Other Diseases
This section briefly mentions a few other age-diseases which I’ve read a little bit about, but haven’t studied in as much depth. I’ll just give a few comments on how they tie in to the cellular processes discussed later.
Arthritis: arthritis is basically inflammation in the joints. I’ve heard plenty of claims that it’s caused by increasing numbers of senescent cells. This would make sense; senescent cell counts are firmly established to increase with age, and senescent cells secrete inflammatory factors, so put 2 and 2 together. On the other hand, I also heard a high-profile clinical trial based on this hypothesis recently failed; I don’t consider that especially strong evidence, since that could easily be due to something specific to the trial. I don’t know enough to have a strong belief on whether senescent cells are the main factor here, but it’s my most likely current model.
Osteoporosis: calcium regulation goes completely bonkers with age. I’ve looked into this only briefly, and my main conclusion is that it’s a confusing mess and I have no idea what’s going on. The most promising direction I’ve stumbled on was in a physiology class, unrelated to aging, when the professor mentioned that osteoblasts (bone-making cells and a major calcium regulator) are derived from immune cells and respond to inflammatory signals. Given that aging in general tends to involve low-grade chronic inflammation (most likely due to increasing numbers of senescent cells), “calcium regulation goes completely bonkers” seems like the sort of thing which would result.
Cancer: the key requirement for cancer is cells with oncogenic mutations. As usual, increasing cancer rates could be caused in two main ways: increasing production rate of mutations, or decreasing removal rate of mutant cells. There are plausible age-related mechanisms for both of these. On the production side, we’ll later discuss how genomic instability relates to ROS and senescence, and in particular the role of transposons. On the removal side, the immune system weakens with age, likely for the same reasons as everything else weakens with age. Also, in this case it’s plausible that an increase in the production rate of precancerous cells and/or senescent cells would slow the removal rate as well, simply because the immune system has limited capacity.
Cataracts: as with hardening of the vasculature, cataracts can be reversed by aminoguanidine, so the same considerations apply.
Core Intermediates
Now we get to the meaty part. At the microscopic level, there’s a handful of pieces which pop up again and again in age-related diseases:
Oxidative damage to DNA, proteins, fats, etc.
ROS
Senescent cells
Inflammation
Mitochondrial dysfunction
Some of these have obvious connections. For instance, more ROS presumably lead to more oxidative damage to DNA/proteins/fats/etc. Some key questions here:
Where are the ROS produced? Mitochondria are the top candidate—there’s a known mechanism for ROS production by mitochondria, as well as experimental evidence that mitochondrion-targeted antioxidants specifically reduce ROS-induced damage.
How do the ROS and/or damaged molecules move between compartments, e.g. nucleus/cytoplasm/extracellular? I have seen very little on this, and consider it a major blindspot. I’m not sure if it’s a blindspot for the field or if I just haven’t found the right cluster of papers.
Are the quantitative changes in DNA/protein/fat damage compatible with a single underlying cause? Do they match plausible estimates of ROS from dysfunctional mitochondria? Again, I haven’t seen Fermi estimates here, but I’d like to.
We do have evidence (from aminoguanidine and similar drugs; good overview here for protein damage) that ROS are causal for various types of damage.
Another connection: we’ve already mentioned that senescent cells release inflammatory factors, the so-called “senescence-associated secretory phenotype” (SASP). The one question here for which I haven’t seen a clear answer is whether increasing numbers of senescent cells quantitatively match age-related increases in inflammation. Drugs to remove senescent cells are a hot area right now, and should provide more evidence on whether senescent cells are causal for age-related inflammation.
So we have two clusters of probably-causally-connected processes. One of these involves dysfunctional mitochondria producing excess ROS which damages DNA/proteins/fats, and the other involves senescent cells inducing inflammation.
The really big discovery of the past twenty years was the connection between cellular senescence and ROS/mitochondrial dysfunction. Turns out, ROS, DNA damage, and mitochondrial dysfunction are all tied together in one bistable feedback loop, and cellular senescence is basically a state-change in that feedback loop.
The DNA Damage <-> Mitochondrial ROS Feedback Loop Underlying Senescence
The two key papers here are “Feedback between p21 and reactive oxygen production is necessary for cell senescence“ and “Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence”. Another group found basically the same phenomenon, but they found some weird differences compared to normal senescence which I think were probably artifacts of how they blocked mitochondrial function, so they’re less directly relevant to cellular senescence in the wild.
Here’s how the feedback loop works:
A cell’s DNA is damaged, inducing a damage response. (p21 is the main signalling molecule which activates the damage-response genes.)
As part of this damage response, mitochondria are shifted into a lower-efficiency state, producing less energy and more ROS.
The ROS then further damage DNA.
At low levels of activation, the ROS do not produce enough DNA damage to make this loop self-sustaining. The cell stays in a low-ROS, low-damage state—the “normal” state. But once it passes some threshold, the positive feedback takes off, and the cell transitions into a high-ROS, high-damage state—the “senescent” state.
Notably, after a few days, the cell changes in some other (not yet fully understood) manner, locking in the senescent state. After this point, even if ROS are suppressed, the cell will remain in senescence rather than switching back to the normal-cell state. Transposons are one plausible candidate for this lock-in; more on that shortly.
The experimental evidence for this process is beautiful (I definitely recommend those papers, especially the second). And it neatly unifies all of the pieces we listed above: cellular senescence is a positive feedback loop between ROS, damage and mitochondrial dysfunction, and the SASP connects it all to inflammation.
There’s still one big question: why is this positive feedback loop active more often, for more cells, in old age? The whole feedback loop is fast, senescent cells are removed on a timescale of days to weeks, so there must be some upstream change either increasing the rate of triggering senescence (presumably by somehow damaging DNA, possibly via ROS, or inducing mitochondrial dysfunction) or decreasing the rate of removal of senescent cells. In fact, both of these do occur, although personally I think the increase in rate of triggering senescence is much more likely to be causal.
Further Up The Causal Chain
There are lots of stories about how various plausible root causes could, perhaps, trigger the senescence feedback loop. I think transposons are the most likely candidate, though mitochondrial mutations are a plausible mechanism as well. We’ll talk about both of those in the next two subsections. Most other proposed root causes can, I think, be ruled out at this point—we’ll talk about some of those in the “Other Root Causes” subsection below.
Transposons
A transposon is a gene whose main function is to copy itself. The LINE-1 family of transposons (most common active transposons in humans) consist of a protein which snips the DNA, and another protein which reverse-transcribes the transposon’s mRNA into DNA attached to the snipped end. (I’m glossing over some details here.)
These things are extremely common in the genome—more than half of human DNA consists of dead transposons. (“Dead” here means that the transposon mutated at some point in evolutionary history, so it’s no longer functional.) Fortunately, the number of non-dead transposons in the human genome is much smaller - even the highest estimates I’ve seen put the number typically below 100.
We do have mechanisms to repress transposon activity, most notably epigenetic mechanisms. Most DNA is usually tightly coiled up around little cylindrical proteins (called histones), where it can’t be easily transcribed. “Epigenetics” typically refers to modifications of the DNA and/or histones which make the coils tighter or looser, making the DNA difficult or easy to access. Most transposons are epigenetically tagged so that they’re kept tightly coiled most of the time. Indeed, an argument could be made that this is the primary role of epigenetics—it’s certainly what most epigenetic modifications are doing most of the time, since transposons fill so much of the genome.
There’s an obvious story by which transposons could be a root cause of aging. Most of the time they’re repressed, but every once in a while, one of them manages to copy itself. Once it’s copied, there’s no undoing it—the transposon count will only go up over time. Eventually, there’s enough active transposons that the repression mechanisms aren’t so reliable. At that point, the transposon protein which snips the DNA will be expressed quite a bit, resulting in lots of DNA damage—those snips are exactly the sort of damage which can trigger senescence. (Note: it’s a lot easier to snip the DNA than to reverse-transcribe, so I generally expect there to be a lot more snips than successful transposon-copy events.)
Setting aside this root-cause story for a moment, there’s also evidence that cellular senescence causes derepression of transposons. We’ll talk more about the details of that later, but the key idea here is that there’s a trade-off at the cellular level between repairing damage and repressing the transposons. When DNA damage is high, the cell temporarily shifts resources to repair that damage, deregulating the transposons. In senescence, the level of DNA damage is constantly high, so transposons go wild.
Of course, transposons themselves cause DNA damage (i.e. the snips), so eventually this can lock in senescence. That’s my current best understanding of why senescence gets “locked in” after a few days: with transposons driving the DNA damage, the cell will remain senescent even if ROS are suppressed. It’s a second senescence feedback loop, slower to start up, but permanent—once the transposons are active enough, the cell cannot leave senescence.
Unfortunately, this makes it difficult to distinguish the chicken from the egg. We know that senescence can cause transposon activity, but we also suspect that transposon activity comes first and causes senescence. We can’t test that hypothesis just by looking at transposon activity in senescent cells. In principle, we could test it by looking for an age-related increase in transposon count in non-senescent cells, but that turns out to be actually-pretty-difficult in practice. (Modern DNA sequencing involves breaking the DNA into little pieces, sequencing those, then computationally reconstructing which pieces overlap with each other. That’s a lot more difficult when the pieces you’re interested in have millions of near-copies filling most of the genome. Also, the copy-events we’re interested in will vary from cell to cell.)
One more thing to note about this model: suppose that a stem cell has a high transposon count, but not high enough to undergo senescence itself. That stem cell will pump out new cells, as stem cells typically do, and those cells will themselves be close to senescence. We should therefore see little clusters of senescent cells, each derived from one stem cell. This is where I expect most senescent cells come from in aged tissue. The “ragged red” sections of aging muscle cells are a good example—one satellite cell is near senescence, so it pumps out nuclei which rapidly senesce, and the section of the muscle cell near that satellite ends up senescing.
This picture also offers an obvious story for cancer—transposons are a major driver of mutations.
Mitochondrial Mutations
Mitochondrial mutations are the center of the “mitochondrial free radical theory of aging” (MIFRA); Aubrey de Gray’s MIFRA book provides an excellent (though out-of-date) summary of the model. Some parts of the model have been pretty well nailed down by evidence at this point—e.g. the idea that most core diseases of aging are mainly driven by ROS, which in turn are produced by mitochondria. The positive feedback loop underlying senescence even further connects symptoms of aging to mitochondrial ROS.
The main piece of the MIFRA model which still seems up-in-the-air is the idea that the root cause is mitochondrial mutations.
Background: mitochondria have their own separate DNA. There’s only a handful of genes on it; most necessary mRNA/protein sequence is supplied by the nuclear DNA. But the mitochondria’s little DNA is particularly prone to mutation—it doesn’t have the nucleus keeping it safely separated from the highly-energetic processes of the rest of the cell. The flip side is that mitochondria turn over frequently, separate from turnover of the cell itself, and they have quality-control mechanisms—e.g. if a mitochondrion isn’t producing energy (as indicated by low transmembrane potential) then it’s broken down.
A key idea hypothesis for the mitochondrial mutation model is that mutant mitochondria which are only partially defective aren’t broken down. In fact, under this hypothesis, such mitochondria have a replicative advantage, and can expand to take over the whole cell. This cell then becomes a “hotspot”, pumping out lots of ROS.
Today, we would expect that such hotspots are senescent cells—this degree of mitochondrial dysfunction should certainly be enough to induce senescence.
There is some evidence for this—for instance, mitochondrial mutants tend to take over whole cells, rather than being spread evenly across cells. However, there just aren’t very many of them—senescent cells outnumber mutant-mitochondria-dominant cells by a wide margin in old age (order of magnitude: think 10% vs 1%). Also, as we mentioned earlier, senescent cells don’t reproduce and do turn over, so even if mutant mitochondria took over one cell, they’d need to somehow expand to others.
This still doesn’t rule out the model entirely. Some possibilities:
Maybe a small fraction of senescent cells are long-lived, and these produce enough ROS to induce senescence in a much larger population of cells.
Maybe mutant mitochondria spread before the cell is cleared (there is evidence for exchange of mitochondria between neighboring cells).
Maybe stem cells with some mutant mitochondria produce new cells which go on to rapidly senesce, much like we hypothesized for transposons.
On the other hand, it’s also plausible (I think more plausible) that defective mitochondria are negatively selected in healthy cells, and they only expand to take over in already-senescent cells, where all the mitochondria are already pumping out less energy and more ROS anyway.
Other Root Causes?
Telomeres
DNA has repetitive regions called telomeres at the ends . Each time a cell divides, the copying starts a little ways in from the end, so the telomeres get a bit shorter. Eventually, the telomere runs out, and the bare DNA end is interpreted as damage, inducing senescence. This has long been known to cause cellular senescence in vitro, and was hypothesized as a root cause of aging.
Indeed, telomeres are known to shorten with age. On the other hand, upregulating telomerase seems to do approximately nothing to prevent cellular senescence or aging more generally. What’s going on here?
Stem cells produce a protein (telomerase) which extends their telomeres. Since most cells are replaced regularly by stem cells, that should be enough to prevent telomere-induced senescence. But then why do we observe shorter telomeres in old age? The key result here is that DNA damage, and ROS in particular, shorten telomeres. In vivo, telomere length is mainly a measure of ROS damage, not a measure of age directly. So, while telomere loss can cause senescence, the main thing which causes telomere loss is not gradual shortening as cells divide, but rather ROS-induced damage.
So, telomere loss is likely involved as an intermediate cause (as a type of DNA damage induced by ROS), but not a root cause.
AGEs
Advanced glycation end-products (AGEs) are proteins which have somewhat-randomly reacted with a sugar in a Maillard reaction—the same type of reaction which browns foods and gives them flavor when cooking. These products are hypothesized to accumulate long-term in the body, since they can’t be broken down.
I don’t know whether there’s any evidence that these molecules actually accumulate long-term. (Just because they’re not broken down doesn’t mean they’re not simply excreted.) I haven’t seen direct evidence, but I haven’t searched very carefully either, and I haven’t seen direct evidence against.
The main argument I’ve seen in favor of AGEs as a root cause of (some) diseases of aging was from aminoguanidine. It seems like people thought it prevented AGE formation before they realized that it interrupted oxidative damage more broadly, and thus interpreted results from aminoguanidine as indicative of a causal role for AGEs. In hindsight, even if this had shown a causal role for AGEs, it would have ruled them out as a root cause: the effects of AG rapidly wear off once administration of the drug ceases, so it’s definitely not blocking any root cause.
Senescent Cells as Root Cause
For a while, people hypothesized that senescent cells accumulate with age without turning over, acting as a root cause. As mentioned earlier, the actual evidence suggests that senescent cells turn over on a timescale of days to weeks, which would mean this theory is wrong—senescent cell accumulation is not a root cause.
However, there is a saving throw: maybe a small subset of senescent cells are longer-lived, and the experiments measuring senescent cell turnover time just weren’t capturing the long-lived subset in particular. Results from senolytics (drugs which kill senescent cells) suggest this is also wrong: the effects of senolytics rapidly wear off once the drug stops being administered, whereas reversing a root cause should set an organism back to a youthful state longer-term.
Protein Damage, DNA Damage, Etc as Root Cause
Sometimes people suggest protein damage, DNA damage, etc, as root causes. These generally turn over on fast timescales, so… no. I expect that most of these people do not have any thought-out notion of what “root” means in “root cause”, and are just using it as a synonym for “really important”.
Other Pieces of The Pathway
This last section will briefly dive into a couple other aspects of the core pathways of aging which I expect people might be interested in, and talk about how they fit into the picture.
Sirtuins and NAD
David Sinclair published a popular book on aging a couple years ago, mainly talking about his own research areas. The sirtuins are one of the main key pieces of that research.
We mentioned earlier that, when damage is detected, the cell redirects resources from repressing transposons to repairing damage. Sirtuins are one such resource. They directly trade off genomic stability (including transposon repression) for repair capacity.
Notably, sirtuins consume the energy carrier NAD as part of their repair role. Lots of things use NAD as an energy carrier, switching it between a high-energy and low-energy state, but sirtuins actually consume it—the whole molecule is incorporated into a new structure. Usually the cell has rather a lot of NAD, but if the damage load is high and the sirtuins are doing lots of repairs, then it can be depleted. That leaves less of it for various cellular processes which use NAD as an energy carrier—including mitochondrial energy production.
This all fits neatly into our main model: high ROS-induced damage draws sirtuins away from transposon repression, so they become active. Meanwhile, the sirtuins’ consumption of NAD can interfere with mitochondrial function, resulting in more ROS production.
This also adds in one more piece: at younger ages, certain kinds of cellular stress (like radiation exposure or chemical damage from an inflammatory response to an infection) can also damage DNA, temporarily reducing repression of transposons. This probably won’t result in immediate senescence in most cells, but a few transposons may copy before everything goes back to normal. The aging clock ticks forward a little faster than usual, due to these events.
Damaged Proteins Connect To Everything
We’ve mentioned oxidatively damaged proteins many times now. What we didn’t mention was the numbers: in young organisms, perhaps 10% of protein is damaged. In old organisms, it’s more like 20-30%.
That is a percentage of all proteins. And almost everything our cells do is done by proteins.
Natural conclusion: efficiency of a very wide variety of processes will be thrown off in aging.
In most cases, this shouldn’t be too noticeable—we’re only talking about a 10-20% change, and random noise does that for most protein species anyway. And the body has lots of feedback loops in place to handle exactly this sort of thing. By and large, biological networks evolve to be robust to 10-20% changes in protein concentrations. But, it does make things difficult for science: if there’s a 10-20% change in everything, then there are always going to be statistically significant age-related changes in everything, even though most of them aren’t actually all that relevant.
So, be warned: there’s lots of 10-20% age-related changes all over the place which mostly aren’t that relevant.
Recap
Here’s the core positive feedback loop again:
A cell’s DNA is damaged, inducing a damage response.
As part of this damage response, mitochondria are shifted into a lower-efficiency state, producing less energy and more ROS.
The ROS then further damage DNA.
Since this is a positive feedback loop, it has two stable states: one state with low damage and ROS (the “normal” cell state), and one with high damage and ROS (the “senescent” cell state).
A few days after senescence is triggered, the cell’s transposons become active, copying themselves and further damaging the DNA. At this point, the transposon activity alone is enough to maintain the senescent state, and senescence is locked in. The cell will remain senescent until it’s cleared out by the immune system, a few days to weeks later.
What causes more senescent cells as we age? The transposon model says that transposon count increases with age—they are the root cause which permanently accumulates over time. Once the transposon count in a cell is high enough, it produces enough damage to trigger the senescence feedback loop. More specifically, we end up with stem cells with enough transposons to be just below the senescence trigger, and these stem cells produce new cells which rapidly senesce.
Senescent cells release inflammatory factors (the SASP) as well as ROS. These cause the bulk of age-related diseases. ROS damage to fats causes buildup of fatty streaks and eventually plaques in the arteries—i.e. atherosclerosis—eventually leading to blockage and strokes. Damage to proteins hardens the blood vessels, leading to heart failure and aneurysm. Chronic inflammation underlies arthritis and possibly osteoporosis. Senescence itself also leads to loss of cells, including muscle loss.
Finally, in very old age, the whole process can accelerate: ROS produced by senescent cells can cause damage in adjacent cells, inching them closer to senescence as well. The more cells senesce, the more damage they deal to healthy cells. Eventually the whole thing goes supercritical (though on a slow timescale), leading to an exponential acceleration of disease progression in old age.
- We Choose To Align AI by (1 Jan 2022 20:06 UTC; 286 points)
- That’s Not How Epigenetic Modifications Work by (24 May 2025 0:15 UTC; 68 points)
- Voting Results for the 2021 Review by (1 Feb 2023 8:02 UTC; 66 points)
- On the Evolvability of Biological Immortality by (28 Jul 2025 21:13 UTC; 20 points)
- Hypothesis: lab mice have more active transposons then wild mice by (5 Jun 2021 12:36 UTC; 20 points)
- Should we vaccinate against PGBD5 which codes for a transposase? by (8 Jun 2021 19:22 UTC; 18 points)
- 's comment on A primer on the current state of longevity research by (22 Aug 2024 19:41 UTC; 13 points)
- 's comment on How To Write Quickly While Maintaining Epistemic Rigor by (2 Sep 2021 17:52 UTC; 10 points)
- 's comment on The Efficient Market Hypothesis in Research by (8 Jul 2021 21:21 UTC; 10 points)
- 's comment on The aducanumab approval by (17 Jun 2021 14:16 UTC; 5 points)
- 's comment on Open & Welcome Thread September 2021 by (5 Jan 2022 0:25 UTC; 5 points)
- 's comment on Have you considered getting rid of death? by (31 Aug 2022 16:17 UTC; 2 points)
- 's comment on ChristianKl’s Shortform by (10 Jun 2021 22:35 UTC; 2 points)
- 's comment on What can we learn from traditional societies? by (5 Oct 2021 12:13 UTC; 2 points)
- 's comment on Linch’s Shortform by (31 Mar 2026 13:17 UTC; 2 points)
- 's comment on Have you considered getting rid of death? by (14 Sep 2022 0:39 UTC; 1 point)

Both this document and John himself have been useful resources to me as I launch into my own career studying aging in graduate school. One thing I think would have been really helpful here are more thorough citations and sourcing. It’s hard to follow John’s points (“In sarcopenia, one cross-section of the long muscle cell will fail first—a “ragged red” section—and then failure gradually spreads along the length.”) and trace them back to any specific source, and it’s also hard to know which of the synthetic insights are original to John and which are insights from the wider literature that John is echoing here.
While eschewing citations makes the post a little easier to scan, and probably made it a lot easier to write, I think that it runs the risk of divorcing the post from the wider literature and making it harder for the reader to relate this blog post to the academic publications it is clearly drawing upon. It would have also been helpful if John had more often referenced specific terms—when he says “Modern DNA sequencing involves breaking the DNA into little pieces, sequencing those, then computationally reconstructing which pieces overlap with each other,” it’s true, but also, DNA sequencing methods are diverse and continue to evolve on a technological level at a rapid pace. It’s hard to know exactly which set of sequencing techniques he had in mind, or how much care he took in making sure that there’s no tractable way to go about this.
Overall, I’m just not sure to what extent I ought to let this post inform my understanding of aging, as opposed to inspiring and motivating my research elsewhere. But I still appreciate John for writing it—it has been a great launch point.