Significantly Enhancing Adult Intelligence With Gene Editing May Be Possible
In the course of my life, there have been a handful of times I discovered an idea that changed the way I thought about where our species is headed. The first occurred when I picked up Nick Bostrom’s book “superintelligence” and realized that AI would utterly transform the world. The second was when I learned about embryo selection and how it could change future generations. And the third happened a few months ago when I read a message from a friend of mine on Discord about editing the genome of a living person.
We’ve had gene therapy to treat cancer and single gene disorders for decades. But the process involved in making such changes to the cells of a living person is excruciating and extremely expensive. CAR T-cell therapy, a treatment for certain types of cancer, requires the removal of white blood cells via IV, genetic modification of those cells outside the body, culturing of the modified cells, chemotherapy to kill off most of the remaining unmodified cells in the body, and reinjection of the genetically engineered ones. The price is $500,000 to $1,000,000.
And it only adds a single gene.
This is a big problem if you care about anything besides monogenic diseases. Most traits, like personality, diabetes risk, and intelligence are controlled by hundreds to tens of thousands of genes, each of which exerts a tiny influence. If you want to significantly affect someone’s risk of developing lung cancer, you probably need to make hundreds of edits.
If changing one gene to treat cancer requires the involved process described above, what kind of tortuous process would be required to modify hundreds?
It seemed impossible. So after first thinking of the idea well over a year ago, I shelved it without much consideration. My attention turned to what seemed like more practical ideas such as embryo selection.
Months later, when I read a message from my friend Kman on discord talking about gene editing adults in adult brains, I was fairly skeptical it was worth my time.
How could we get editing agents into all 200 billion brain cells? Wouldn’t it cause major issues if some cells received edits and others didn’t? What if the gene editing tool targeted the wrong gene? What if it provoked some kind of immune response?
But recent progress in AI had made me think we might not have much time left before AGI, so given that adult gene editing might have an impact on a much shorter time scale than embryo selection, I decided it was at least worth a look.
So I started reading. Kman and I pored over papers on base editors and prime editors and in-vivo delivery of CRISPR proteins via adeno-associated viruses trying to figure out whether this pipe dream was something more. And after a couple of months of work, I have become convinced that there are no known fundamental barriers that would prevent us from doing this.
There are still unknowns which need to be worked on. But many pieces of a viable editing protocol have already been demonstrated independently in one research paper or another.
If this works, it’s going to be a very big deal
It is hard to overstate just how big a deal it would be if we could edit thousands of genes in cells all over the human body. Nearly every trait and every disease you’ve ever heard of has a significant genetic component, from intelligence to breast cancer. And the amount of variance already present in the human gene pool is stunning. For many traits, we could take someone from the 3rd percentile to the 97th percentile by editing just 20% of the genes involved in determining that trait.
Tweaking even a dozen genes might be able to halt the progression of Alzheimer’s, or alleviate the symptoms of major autoimmune diseases.
The same could apply to other major causes of aging: diabetes, heart disease, cancers. All have genetic roots to some degree or other. And all of this could potentially be done in people who have already been born.
Of particular interest to me is whether we could modify intelligence. In the past ten thousand years, humans have progressed mostly through making changes to their tools and environment. But in that time, human genes and human brains have changed little. With limitations to brain power, new impactful ideas naturally become harder to find.
This is particularly concerning because we have a number of extremely important technical challenges facing us today and a very limited number of people with the ability to make progress on them. Some may simply be beyond the capabilities of current humans.
Of these, the one I think about most is technical AI alignment work. It is not an exaggeration to say that the lives of literally everyone depend on whether a few hundred engineers and mathematicians can figure out how to control the machines built by the mad scientists in the office next door.
Demis Hassabis has a famous phrase he uses to describe his master plan for Deepmind: solve intelligence, then use that intelligence to solve everything else. I have a slightly more meta level plan to make sure Demis’s plan doesn’t kill everyone; make alignment researchers smarter, then kindly ask those researchers to solve alignment.
This is quite a grandiose plan for someone writing a LessWrong blog post. And there’s a decent chance it won’t work out for either technical reasons or because I can’t find the resources and talent to help me solve all the technical challenges. At the end of the day, whether or not anything I say has an impact will depend on whether I or someone else can develop a working protocol.
How does intelligence even work at a genetic level?
Our best estimate based on the last decade of data is that the genetic component of intelligence is controlled by somewhere between 10,000 and 24,000 variants. We also know that each one, on average, contributes about +-0.2 IQ points.
Genetically altering IQ is more or less about flipping a sufficient number of IQ-decreasing variants to their IQ-increasing counterparts. This sounds overly simplified, but it’s surprisingly accurate; most of the variance in the genome is linear in nature, by which I mean the effect of a gene doesn’t usually depend on which other genes are present.
So modeling a continuous trait like intelligence is actually extremely straightforward: you simply add the effects of the IQ-increasing alleles to to those of the IQ-decreasing alleles and then normalize the score relative to some reference group.
To simulate the effects of editing on intelligence, we’ve built a model based on summary statistics from UK Biobank, the assumptions behind which you can find in the appendix
Based on the model, we can come to a surprising conclusion: there is enough genetic variance in the human population to create a genome with a predicted IQ of about 900. I don’t expect such an IQ to actually result from flipping all IQ-decreasing alleles to their IQ-increasing variants for the same reason I don’t expect to reach the moon by climbing a very tall ladder; at some point, the simple linear model will break down.
But we have strong evidence that such models function quite well within the current human range, and likely somewhat beyond it. So we should actually be able to genetically engineer people with greater cognitive abilities than anyone who’s ever lived, and do so without necessarily making any great trade-offs.
Even if a majority of iq-increasing genetic variants had some tradeoff such as increasing disease risk (which current evidence suggests they mostly don’t), we could always select the subset that doesn’t produce such effects. After all, we have 800 IQ points worth of variants to choose from!
Total maximum gain
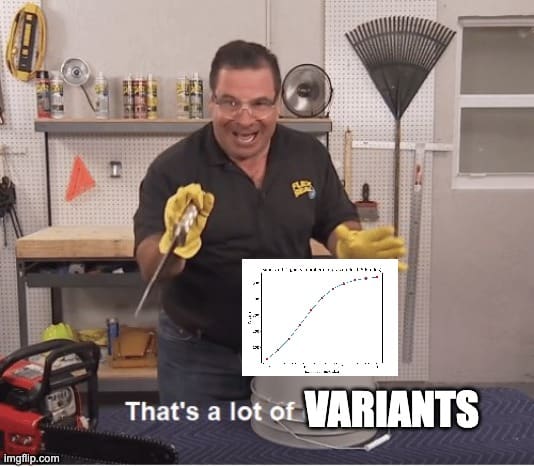
Given the current set of publicly available intelligence-associated variants available in UK biobank, here’s a graph showing the expected effect on IQ from editing genes in adults.
Not very impressive! There are several factors at play deflating the benefits shown by this graph.
As a rough estimate, we expect about 50% of the targeted edits to be successfully made in an average brain cell. The actual amount could be more or less depending on editor and delivery efficiency.
Some genes we know are involved in intelligence will have zero effect if edited in adults because they primarily influence brain development (see here for more details). Though there is substantial uncertainty here, we are assuming that, on average, an intelligence-affecting variant will have only 50% of the effect size as it would if edited in an embryo.
We can only target about 90% of genetic variants with prime editors.
About 98% of intelligence-affecting alleles are in non-protein-coding regions. The remaining 2% of variants are in protein-coding regions and are probably not safe to target with current editing tools due to the risk of frameshift mutations.
Off-target edits, insertions and deletions will probably have some negative effect. As a very rough guess, we can probably maintain 95% of the benefit after the negative effects of these are taken into account.
Most importantly, the current intelligence predictors only identify a small subset of intelligence-affecting genetic variants. With more data we can dramatically increase the benefit from editing for intelligence. The same goes for many other traits such as Alzheimer’s risk or depression.
The limitations in 1-5 together reduce the estimated effect size by 79% compared to making perfect edits in an embryo. If you could make those same edits in an embryo, the gains would top out around 30 IQ points.
But the bigger limitation originates from the size of the data set used to train our predictor. The more data used to train an intelligence predictor, the more of those 20,000 IQ-affecting variants we can identify, and the more certain we can be about exactly which variant among a cluster is actually causing the effect.
And the more edits you can make, the better you can take advantage of that additional data. You can see this demonstrated pretty clearly in the graph below, where each line represents a differently sized training set.
Our current predictors are trained using about 135,000 samples, which would place it just above the lowest line on the graph. There are existing databases right now such as the million veterans project with sample sizes of (you guessed it) one million. The expected gain from editing using a predictor trained on such a data set is shown by the orange line in the graph above.
Companies like 23&Me genotyped their 12 millionth customer two years ago and could probably get at perhaps 3 million customers to take an IQ test or submit SAT scores. A predictor trained with that amount of data would perform about as well as the green line on the graph above.
So larger datasets could increase the effect of editing by as much as 13x! If we hold the number of edits performed constant at 2500, here’s how the expected gain would vary as a function of training set size:
Now we’re talking! If someone made a training set with 3 million data points (a realistic possibility for companies like 23&Me), the IQ gain could plausibly be over 100 points (though keep in mind the uncertainties listed in the numbered list above). Even if we can only make 10% of that many edits, the expected effect size would still be over one standard deviation.
Lastly, just for fun, let’s show the expected effects of 2500 edits if they were made in an embryo and we didn’t have to worry about off-targets or limitations of editors (don’t take this graph too seriously)
The sample size issue could be solved pretty easily by any large government organization interested in collecting intelligence data on people they’ve already genotyped. However, most of them are not currently interested.
Why has it been so hard to get larger sample sizes?
There is a taboo against direct investigation of the genetics of intelligence in academia and government funding agencies. As a result, we have collected a pitifully small amount of data about intelligence, and our predictors are much worse as a result.
So people interested in intelligence instead have to research this proxy trait that is correlated with intelligence called “educational attainment”. Research into educational attainment IS allowed, so we have sample sizes of over 3 million genome samples with educational attainment labels and only about 130,000 for intelligence.

Government agencies have shown they could solve the data problem if they feel so inclined. As an alternative, if someone had a lot of money they could simply buy a direct-to-consumer genetic testing company and either directly ask participants to take an IQ test or ask them what their SAT scores were.
Or someone who wants to do this could just start their own biobank.
How can you even make these edits anyways? Isn’t CRISPR prone to errors?
Another of my initial doubts stemmed from my understanding of gene editing tools like CRISPR, and their tendency to edit the wrong target or even eliminate entire chromosomes.
You see, CRISPR was not designed to be a gene editing tool. It’s part of a bacterial immune system we discovered all the way back in the late 80s and only fully understood in 2012. The fact that it has become such a versatile gene editing tool is a combination of luck and good engineering.
The gene editing part of the CRISPR system is the Cas9 protein, which works like a pair of scissors. It chops DNA in half at a specific location determined by this neat little attachment called a “guide RNA”. Cas9 uses the guide RNA like a mugshot, bumping around the cell’s nucleus until it finds a particular three letter stretch of DNA called the PAM. Once it finds one, it unravels the DNA starting at the PAM and checks to see if the guide RNA forms a complementary match with the exposed DNA.
Cas9 can actually measure how well the guide RNA bonds to the DNA! If the hybridization is strong enough (meaning the base pairs on the guide RNA are complementary to those of the DNA), then the Cas9 “scissors” will cut the DNA a few base pairs to the right of the PAM.
So to replace a sequence of DNA, one must introduce two cuts: one at the start of the DNA and one at the end of it.
Once Cas9 has cut away the part one wishes removed, a repair template must be introduced into the cell. This template is used by the cell’s normal repair processes to fill in the gap.
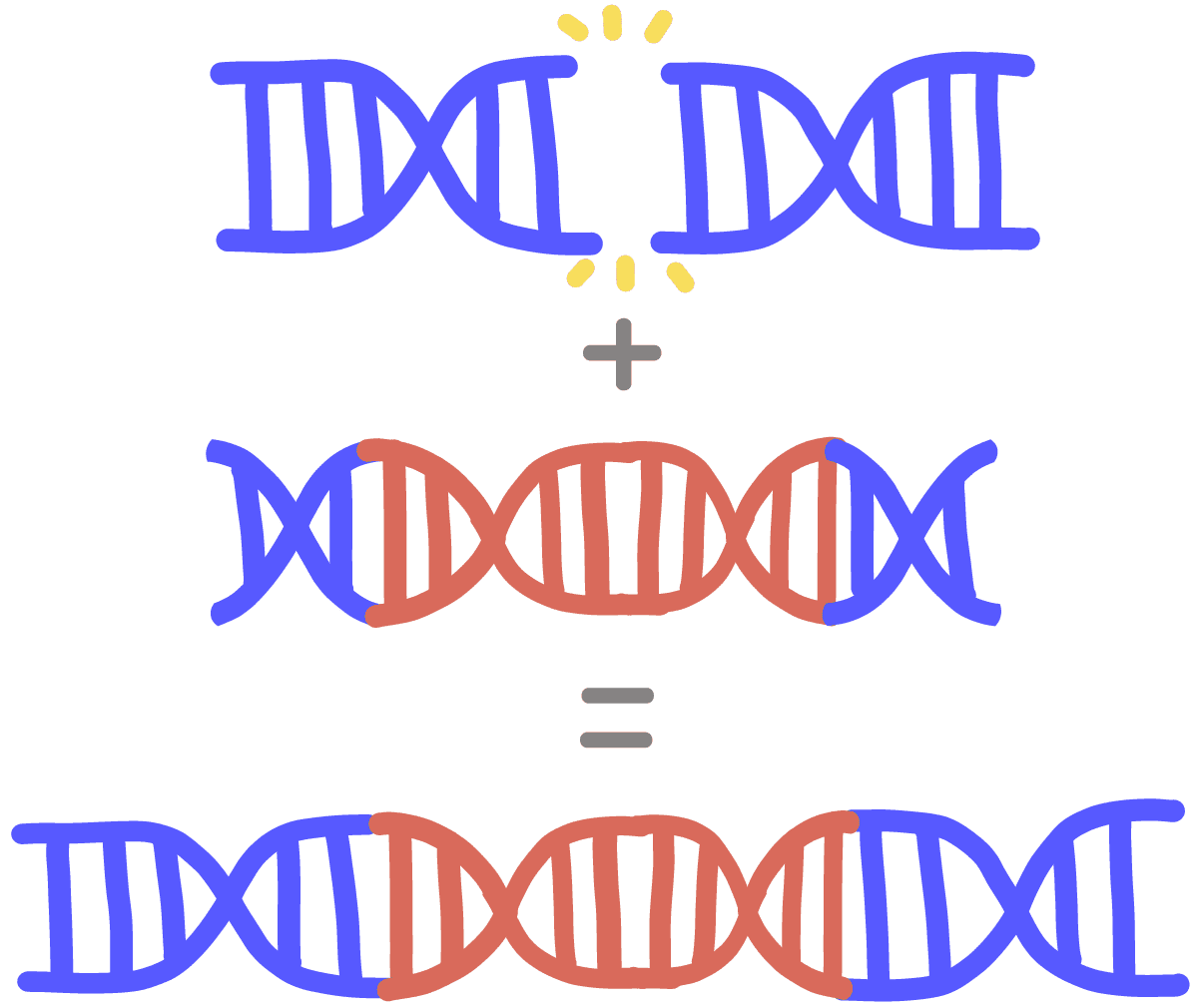
Or at least, that’s how it’s supposed to work. In reality a significant portion of the time the DNA will be stitched back together wrong or not at all, resulting in death of the chromosome.
This is not good news for the cell.
So although CRISPR had an immediate impact on the life sciences after it was introduced in 2012, it was rightfully treated with skepticism in the context of human genome editing, where deleting a chromosome from someone’s genome could result in immediate cell death.
Had nothing changed, I would not be writing this post. But in 2016, a new type of CRISPR was created.
The first base editor
Many of the problems with CRISPR stem from the fact that it cuts BOTH strands of DNA when it makes an edit. A double stranded break is like a loud warning siren broadcast throughout the cell. It can result in a bunch of new base pairs being added or deleted at the break site. Sometimes it even results in the immune system ordering the cell to kill itself.
So when David Liu started thinking about how to modify CRISPR to make it a better gene editor, one of the first things he tried to do was prevent CRISPR from making double-stranded breaks.
He and members of his lab came up with a very creative way of doing this. They modified the Cas9 scissors so that they would still seek out and bind to a matching DNA sequence, but wouldn’t cut the DNA. They then attached a new enzyme to Cas9 called a “deaminase”. Its job is to change a single base through a chemical reaction.
They called this new version of CRISPR a “base editor”
There’s a lot more detail to go into regarding how base editors work which I will explain in another post, but the most noteworthy trait of base editors is how reliable they are in comparison to old school Cas9.
Base editors make the correct edit between 60 and 180 times as often as the original Cas9 CRISPR. They also make 92% fewer off-target edits and 99.7% fewer insertion and deletion errors.
The most effective base editor I’ve found so far is the transformer base editor, which has an editing efficiency of 90% in T cells and no detected off-targets.
But despite these considerable strength, base editors still have two significant weaknesses:
Of the 12 possible base pair swaps, they can only target 6.
With some rare exceptions they can usually only edit 1 base pair at a time.
To address these issues, a few members of the Liu lab began working on a new project: prime editors.
Prime editors; the holy grail of gene editing technology?
Like base editors, prime editors only induce single stranded breaks in DNA, significantly reducing insertion and deletion errors relative to old school CRISPR systems. But unlike base editors, prime editors can change, insert, or delete up to about a hundred base pairs at a time.
The core insight leading to their creation was the discovery that the guide RNA could be extended to serve as a template for DNA modifications.
To greatly oversimplify, a prime editor cuts one of the DNA strands at the edit site and adds new bases to the end of it using an extension of the guide RNA as a template. This creates a bit of an awkward situation because now there are two loose flaps of DNA, neither of which are fully attached to the other strand.
Thankfully the repair enzyme in charge of fixing these types of problems (which are a necessary part of normal cellular function), really likes to cut the unedited flap for reasons I won’t get into. Once it has cut away the other flap and stitched together the edited strand, the situation looks like this:
There’s still one problem left to fix though: the unedited strand is… well… unedited.
To fix this, Liu and Co use another enzyme to cut the bottom strand of DNA near the edit site, which tricks the repair enzymes in charge of fixing mismatches into thinking that the unedited strand has been damaged and needs to be fixed so that it matches the edited one.
So the cell’s own repair enzymes actually complete the edit for us, which is pretty neat if you ask me.
Together, prime editors and base editors give us the power to edit around 90% of the genetic variants we’re interested in with very high precision. Without them, editing genes in a living human body would be infeasible due to off-target changes.
How do you even get editors into brain cells in the first place?
There are roughly 200 billion cells in an average brain. You can’t just use a needle to inject a bunch of gene editing tools into each one. So how do we get them inside?
There are several options available to us including viral proteins, but the delivery platform I like the most is called a lipid nanoparticle. Let me explain the idea.
Dual use technology
Remember COVID vaccines? The first ones released used this new technology called “mRNA”. They worked a little differently from old school immunizations.
The goal of a vaccine is to expose your immune system to part of a virus so that it will recognize and kill it in the future.
One way to do this is by taking a little piece of the virus and injecting it into your body. This is how the Novavax vaccine worked.
Another way is by sending an instruction to MAKE part of the virus to your body’s own cells. This is how the Pfizer and Moderna vaccines worked. And the instruction they used was a little piece of ribonucleic acid called mRNA.
mRNA is an instruction to your cells. If you can get it inside one, the cell’s ribosomes will transcribe mRNA into a protein. It’s a little bit like an executable file for biology.
By itself, mRNA is pretty delicate. The bloodstream is full of nuclease enzymes that will chop them into itty-bitty pieces.
Also, because mRNA basically has root level access to your cells, your body doesn’t just shuttle it around and deliver it like the postal service. That would be a major security hazard.
So to get mRNA inside a cell, COVID vaccines stick them inside a delivery container to protect them from the hostile environment outside cells and to get them where they need to go. This container is a little bubble of fat called a lipid nanoparticle.
If you inject these nanoparticles into your muscle tissue, they will float around for a while before being absorbed by your muscle cells via a process called endocytosis.
Afterwards, the mRNA is turned into a protein by the ribosomes in the muscle cell. In the case of COVID vaccines the protein is a piece of the virus called the “spike” that SARS-CoV-2 uses to enter your tissues.
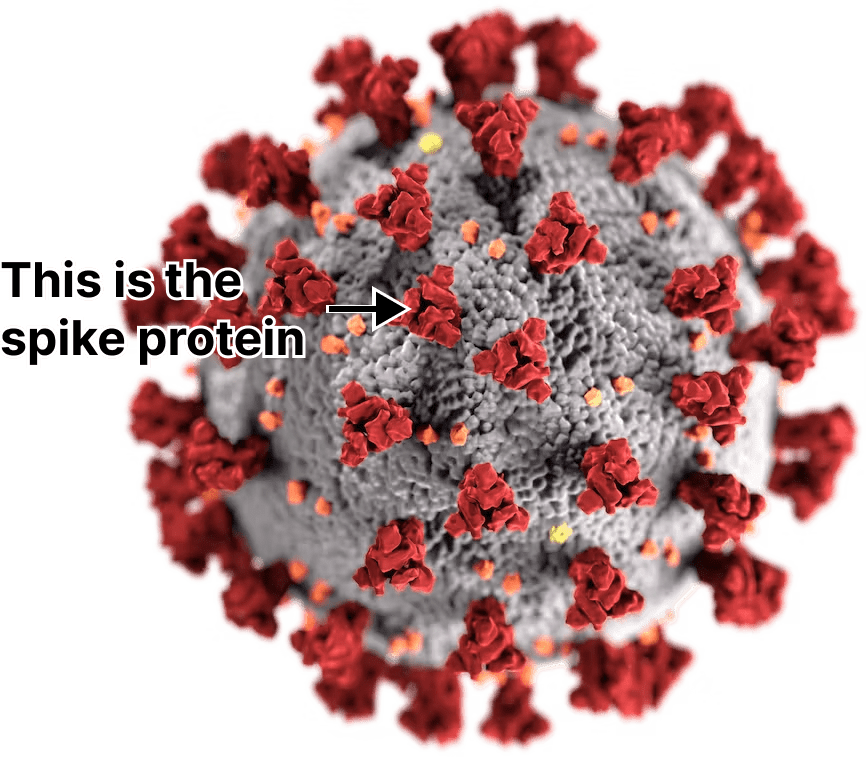
{kind=link}
Once created, some of these spike proteins will be chopped up by enzymes and displayed on the outside of each cell’s membrane. Immune cells patrolling the area will eventually bind to these bits of the spike protein and alert the rest of the immune system, triggering inflammation. This is how vaccines teach your body to fight COVID.
So here’s an idea: what if instead of making a spike protein, we deliver mRNA to make a gene editor?
Instead of manufacturing part of a virus, we’d manufacture a CRISPR-based editing tool like a prime editor. We could also ship the guide RNA in the same lipid nanoparticle, which would bond to the newly created editor proteins, allowing them to identify an editing target.
A nuclear localization sequence on the editor would allow the it to enter the cell’s nucleus where the edit could be performed.
So you inject lipid nanoparticles into the brain?
Not exactly. Brains are much more delicate than muscles, so we can’t just copy the administration method from COVID vaccines and inject nanoparticles directly into the brain with a needle.
Well actually we could try. And I plan to look into this during the mouse study phase of the project in case the negative effects are smaller than I imagine.
But apart from issues with possibly damaging brain or spinal tissue (which we could perhaps avoid), the biggest concern with directly injecting editors into the cerebrospinal fluid is distribution. We need to get the editor to all, or at least most cells in the brain or else the effect will be negligible.
If you just inject a needle into a brain, the edits are unlikely to be evenly distributed; most of the cells that get edited will be close to the injection site. So we probably need a better way to distribute lipid nanoparticles evenly throughout the brain.
Thankfully, the bloodstream has already solved this problem for us: it is very good at distributing resources like oxygen and nutrients evenly to all cells in the brain. So the plan is to inject a lipid-nanoparticle containing solution into the patient’s veins, which will flow through the bloodstream and eventually to the brain.
There are two additional roadblocks: getting the nanoparticles past the liver and blood brain barrier.
By default, if you inject lipid nanoparticles into the bloodstream, most of them will get stuck in the liver. This is nice if you want to target a genetic disease in that particular organ (and indeed, there have been studies that did exactly that, treating monogenic high cholesterol in macaques). But if you want to treat anything besides liver conditions then it’s a problem.
From my reading so far, it sounds like nanoparticles circulate through the whole bloodstream many times, and each time some percentage of them get stuck in the liver. So the main strategy scientists have deployed to avoid accumulation is to get them into the target organ or tissue as quickly as possible.
Here’s a paper in which the authors do exactly that for the brain. They attach a peptide called “angiopep-2” onto the outside of the nanoparticles, allowing them to bypass the blood brain barrier using the LRP1 receptor. This receptor is normally used by LDL cholesterol, which is too big to passively diffuse through the endothelial cells lining the brain. But the angiopep-2 molecule acts like a key, allowing the nanoparticles to use the same door.
The paper was specifically examining this for delivery of chemotherapy drugs to glial cells, so it’s possible we’d either need to attach additional targeting ligands to the surface of the nanoparticles to allow them to get inside neurons, or that we’d need to use another ligand besides angiopep. But this seems like a problem that is solvable with more research.
Or maybe you can just YOLO the nanoparticles directly into someone’s brain with a needle. Who knows? This is why research will need to start with cell culture experiments and (if things go well) animal testing.
What if administration via lipid nanoparticles doesn’t work?
If lipid nanoparticles don’t work, there are two alternative delivery vectors we can look into: adeno-associated viruses (AAVs) and engineered virus-like particles (eVLPs).
AAVs have become the go-to tool for gene therapy, mostly because viruses are just very good at getting into cells. The reason we don’t plan to use AAVs by default mostly comes down to issues with repeat dosing.
As one might expect, the immune system is not a big fan of viruses. So when you deliver DNA for a gene editor with an AAV, the viral proteins often trigger an adaptive immune response. This means that when you next try to deliver a payload with the same AAV, antibodies created during the first dose will bind to and destroy most of them.
There ARE potential ways around this, such as using a different version of an adenovirus with different epitopes which won’t be detected by antibodies. And there is ongoing work at the moment to optimize the viral proteins to provoke less of an immune response. But by my understanding this is still quite difficult and you may not have many chances to redose.
But there are other viral delivery vectors
Engineered virus-like particles, or eVLPs for short, are another way to get gene editors into cells. Like AAVs, they are constructed of various viral proteins and a payload.
But unlike AAVs, eVLPs usually contain proteins, not DNA. This is important because, once they enter a cell, proteins have a much shorter half-life than DNA. A piece of DNA can last for weeks or sometimes even months inside the nucleus. As long as it is around, enzymes will crank out new editor proteins using that DNA template.
Since most edits are made shortly after the first editors are synthesized, creating new ones for weeks is pointless. At best they will do nothing and at worst they will edit other genes that we don’t want changed. eVLPs fix this problem by delivering the editor proteins directly instead of instructions to make them.
They can also be customized to a surprising degree. The yellow-colored envelope glycoprotein shown in the diagram above can be changed to alter which cells take up the particles. So you can target them to the liver, or the lungs, or the brain, or any number of other tissues within the body.
Like AAVs, eVLPs have issues with immunogenicity. This makes redosing a potential concern, though it’s possible there are ways to ameliorate these risks.
Would edits to the adult brain even do anything?
Apart from the technical issues with making edits, the biggest uncertainty in this proposal is how large of an effect we should expect from modifying genes in adults. All the genetic studies on which our understanding of genes is based assume genes differ FROM BIRTH.
We know that some genes are only active in the womb, or in childhood, which should make us very skeptical that editing them would have an effect. For example, here’s a graph showing the relative level of expression of DCX, a gene active in neuronal progenitor cells and migrating immature neurons.
You can see the expression levels peak in the second trimester before dropping by a factor of ~80 by age 27.
We would therefore expect that any changes to promoters or inhibitors of the DCX gene would have little or no effect were they to be made in adulthood.
However, not every gene is like this. Here’s another graph from the same paper showing the relative expression of groups of genes in the hippocampus:
The red, blue and yellow lines, representing the expression of a group of synapse, dendrite, and myelination genes respectively, climb steadily until shortly after birth, then plateau throughout the rest of adulthood. So we could reasonably expect any changes to these genes to have a much stronger effect relative to changes to DEX.
This is one way to develop a basic prior about which genes are likely to have an effect if edited in adults: the size of the effect of a given gene at any given time is likely proportional to its level of expression. Genes that are only active during development probably won’t have as large of an effect when edited.
The paper I linked above collected information about the entire exome, which means we should be able to use its data to form a prior on the odds that editing any given variant will have an effect, though as usual there are some complications with this approach which I address in the appendix.
A systematic analysis of the levels of expressions of all genes throughout the lifespan is beyond the scope of this post, but beyond knowing some proteins reach peak levels in adulthood, here are a few more reasons why I still expect editing to have a large effect:
We know that about 84% of all genes are expressed somewhere in the adult brain. And while almost none of the intelligence-affecting variants are in protein-coding regions (see the section below), they are almost certainly in regions that AFFECT protein expression, such as promoters and inhibitors. One would naturally expect that modifying such protein expression would have an effect on intelligence.
Gene therapy in adults ACTUALLY WORKS. We now have several examples of diseases, such as sickle cell anemia, being completely cured in adults through some form of gene therapy.
We have dozens of examples of traits and diseases whose state is modified by the level of expression of various proteins: Diabetes, Alzheimer’s, autoimmune conditions, skin pigmentation, metabolism, and many others. Since gene editing is ultimately modifying the amount and timing of various proteins, we should expect it to have an effect.
We know the adult brain can change via forming new connections and changing their strengths. That’s how people learn after all!
What if your editor targets the wrong thing?
Though base editors and prime editors have much lower rates of off-target editing than original CRISPR Cas9, off-target editing is still a concern.
There’s a particularly dangerous kind of off-target edit called a frameshift mutation that we really want to avoid. A frameshift mutation happens when you delete (or insert) a number of base pairs that is not a multiple of 3 from a protein-forming region.
Because base pairs are converted into amino acids three at a time, inserting or deleting a letter changes the meaning of all the base pairs that come after the mutation site.
This pretty much always results in a broken protein.
So to minimize the consequences of off-target mutations, we’d do better to target non-protein-coding regions. We already know quite well which regions of the genome are synthesized into proteins. And fortunately, it appears that 98% of intelligence-affecting variants are located in NON-CODING regions. So we should have plenty of variants to target.
What about off-target edits in non-coding regions?
Most of the insertions, deletions and disease-causing variants with small effects are in non-coding regions of DNA. This gives us some evidence that off-target edits made to these regions will not necessarily be catastrophic.
As long as we can avoid catastrophic off-target edits, a few off-targets will be ok so long as they are outweighed by edits with positive effects.
Part of the challenge here is characterizing off-target edits and how they are affected by choice of edit target. If your guide RNA’s binding domain ALMOST matches that of a really critical protein, you may want to think twice before making the edit. Whereas if it’s not particularly close to any target, you probably don’t need to worry as much.
However, it’s still unclear to me how big of a deal off-target edits and indels will be. If you unintentionally insert a few base pairs into the promoter region of some protein, will the promoter just work a little less well, or will it break altogether? I wasn’t able to find many answers in the research literature, so consider this a topic for future research.
It is worth pointing out that long-lived cells in the body such as neurons experience a thousand or more random mutations by age 40 and that, with the exception of cancer, these mutations rarely seem to cause serious dysfunction.
How about mosaicism?
Another potential issue with producing a large number of edits to cells in the brain is cellular mosaicism. The determination of which cell gets which set of edits is somewhat random. Maybe some cells are a little too far away from a blood vessel. Maybe some just don’t get enough of the editor. Regardless of the reason, it’s likely different cells within the brain will receive different sets of edits. Will this cause problems?
It’s possible it will. But as mentioned in the previous section, neurons in the adult brain already have significant genetic differences and most of people seem functional despite these variations. Neurons accumulate about 20-40 single letter mutations per year, with the exact amount varying by brain region and by individual.
By age 45 or so, the average neuron in an adult brain has about 1500 random genetic mutations. And the exact set of mutations each neuron has is somewhat random.
There are likely to be some differences between mutations caused by gene editing and those caused by cellular metabolism, pollutants, and cosmic rays. In particular, those caused by gene editing are more likely to occur at or near functionally important regions.
Still, it should be at least somewhat comforting that neurons usually remain functional when they have a few thousand genetic differences. But it is possible my optimism is misplaced here and mosaicism will turn out to be a significant challenge for some reason I can’t yet foresee. This is yet another reason to conduct animal testing.
Can we make enough edits in a single cell to have a significant effect?
Making a large enough number of edits to have a significant effect on a phenotype of interest is one of the main challenges with this research protocol. I’ve found fairly recent papers such as Qichen & Xue, that show a reasonably large number of edits can be made in the same cell at the same time; 31 edits using base editors and 3 using prime editors. As with nearly all research papers in this field, they used HEK293T cells, which are unusually easy to transfect with editors. We can therefore treat this as an upper bound on the number of simultaneous edits that can be made with current techniques.
Here’s another research paper from Ni et al. where the researchers used prime editors to make 8 simultaneous edits in regenerated wheat plants.
I also know of a PHD student in George Church’s lab that was able to make several thousand edits in the same cell at the same time by targeting a gene that has several thousand copies spread throughout the genome.
So people are trying multiplex editing, but if anyone has tried to make hundreds of edits at a time to different genes, they don’t seem to have published their results. This is what we need to be able to do for in-vivo multiplex editing to have a big impact on polygenic traits and diseases.
However, we don’t necessarily need to introduce all edits with a single injection. If the delivery vector and the editing process doesn’t provoke an adaptive immune response, or if immunosuppressants can be safely used many times, we can just do multiple rounds of administration; 100 edits during the first round, another 100 in the second round and so on.
If there is an immune response, there are some plausible techniques to avoid adverse reactions during subsequent doses, such as using alternative versions of CRISPR with a different antigenic profile. There are a limited number of these alternative CRISPRs, so this technique is not a general purpose solution.
Making hundreds of edits in cultured cells is the first thing I’d like to focus on. I’ve laid out my plans in more detail in “How do we make this real?”
Why hasn’t someone already done this?
There are hundreds of billions of dollars to be made if you can develop a treatment for brain disorders like Alzheimers, dementia and depression. There’s probably trillions to be made if you can enhance human intelligence.
And yet the people who have developed these tools seem to only be using them to treat rare monogenic disorders. Why?
Part of the answer is that “people are looking into it, but in a pretty limited capacity.” For example, here’s a paper that looked at multiplex editing of immune cells in the context of immunotherapy for cancer. And here’s a post that is strikingly similar to many of the ideas I’ve raised here. I only learned of its existence a week before publishing, but have spoken with the authors who had similar ideas about the challenges in the context of their focus on gene therapy for polygenic disease.
Apart from that I can’t find anything. There are 17 clinical trials involving some kind of CRISPR-based gene editing and I can’t find a single one that targets multiple genes.
So here I’m going to speculate on a couple of reasons why there seems to be so little interest in in-vivo multiplex editing.
Black box biology
To believe multiplex editing can work you have to believe in the power of black box biology. Unlike monogenic diseases, we don’t always understand the mechanism through which genetic variants involved in diabetes or intelligence cause their effects.
You have to believe that it is possible to affect a trait by changing genes whose effect you understand but whose mechanism of action you don’t. And what’s more, you need to believe you can change hundreds of genes in a large number of cells in this manner.
To most people, this approach sounds crazy, unpredictable, and dangerous. But the fact is nature mixes and matches combinations of disease affecting alleles all the time and it works out fine in most cases. There are additional challenges with editing such as off-targets and editing efficiency, but these are potentially surmountable issues.
Vague association with eugenics make some academics shy away
A lot of people in Academia are simply very uncomfortable with the idea that genes can influence outcomes in any context other than extremely simple areas like monogenic diseases. I recently attended a conference on polygenic embryo screening in Boston and the amount of denialism about the role of genetics in things as simple as disease outcomes was truly a sight to behold.
In one particularly memorable speech, Erik Turkheimer, a behavioral geneticist from University of Virginia claimed that, contrary to the findings of virtually every published study, adding more data to the training set for genetic predictors simply made them perform worse. As evidence to support this conclusion, he cited a university study on alcohol abuse with a sample size of 7000 that found no associations of genome-wide significance.
Turkheimer neglected to mention that no one could reasonably expect the study to unearth such associations because it was too underpowered to detect all but the most extreme effects. Most studies in this field have 30-100x the number of participants.
In many cases, arguing against these unsupported viewpoints is almost impossible. Genomic Prediction has published about a dozen papers on polygenic embryo selection, many of which are extremely well written and contain novel findings with significant clinical relevance. Virtually no one pays attention. Even scientists writing about the exact same topics fail to cite their work.
Many of them simply do not care what is true. They view themselves as warriors in a holy battle, and they already know anyone talking about genetic improvement is an evil racist eugenicist.
You can convince an open-minded person in a one-on-one discussion because the benefits are so gigantic and the goal is consistent with widely shared values as expressed in hundreds of other contexts.
But a large portion of Academics will just hear someone say “genes matter and could be improved”, think “isn’t that what Hitler did?”, and become forever unreachable. Even if you explain to them that no, Hitler’s theory of genetics was based on a corrupted version of purity ethics and that in fact a lot of his programs were probably dysgenic, and that even if they weren’t we would never do that because it’s evil, it’s too late. They have already passed behind the veil of ignorance. The fight was lost the moment the association with Hitler entered their mind.
Though these attitudes don’t completely prevent work on gene therapies, I think it likely that the hostility towards altering outcomes through genetic means drives otherwise qualified researchers away from this line of work.
Maybe there’s some kind of technical roadblock I’m missing
I’ve now talked to some pretty well-qualified bio PHDs with expertise in stem cells, gene therapy, and genetics. While many of them were skeptical, none of them could point to any part of the proposed treatment process that definitely won’t work. The two areas they were most skeptical of was the ability to deliver an editing vector to the brain and the ability to perform many edits in the same cell. But both seem addressable. Delivery for one, may be solved by other scientists trying to deliver gene editors to the brain to target monogenic conditions like Huntington’s.
There are unknowns of course, and many challenges which could individually or in conjunction make the project infeasible. For example either of the challenges listed above, or perhaps mosaicism is a bigger deal than I expect it to be.
But I’ve only talked to a couple of people. Maybe someone I haven’t spoken with knows of a hard blocker I am unaware of.
People are busy picking lower hanging fruit
It’s a lot easier to target a specific gene that causes a particular disease than it is to target hundreds or thousands of genes which contribute to disease risk. Given prime editors were only created four years ago and base editors only seven, maybe the people with the relevant expertise are just busy picking lower hanging fruit which has a much better chance of resulting in a money-making therapy. The lowest hanging fruit is curing monogenic diseases, and that’s currently where all the focus is.
The market is erring
Some types of markets are highly efficient. Commodities and stocks, for example, are very efficient because a large number of people have access to the most relevant information and arbitrage can happen extremely quickly.
But the more time I’ve spent in private business, and startups in particular, the more I’ve realized that most industries are not as efficient as the stock market. They are far less open, the knowledge is more concentrated, the entry costs are higher, and there just aren’t that many people with the abilities needed to make the markets more efficient.
In the case of multiplex editing, there are many factors at play which one would reasonably expect to result in a less efficient market:
The idea of polygenic editing is not intuitive to most people, even some geneticists. One must either hear it or read about it online.
One must understand polygenic scores and ignore the reams of (mostly) politically motivated articles attempting to show they aren’t real or aren’t predictive of outcomes.
One must understand gene editing tools and delivery vectors well enough to know what is technically possible.
One must believe that the known technical roadblocks can be overcome.
One must be willing to take a gamble that known unknowns and unknown unknowns will not make this impossible (though they may do so)
How many people in the world satisfy all five of those criteria? My guess is not many. So despite how absurd it sounds to say that the pharmaceutical industry is overlooking a potential trillion dollar opportunity, I think there is a real possibility they are. Base editors have been around for seven years and in that time the number of papers published on multiplex editing in human cells for anything besides cancer treatment has remained in the single digits.
I don’t think this is a case of “just wait for the market to do it”.
Can we accelerate development of this technology?
After all that exposition, let me summarize where the work I think will be done by the market and that which I expect them to ignore.
Problems I expect the market to work on
Targeting editors towards specific organs. There are many monogenic conditions that primarily affect the brain, such as Huntington’s disease or monogenic Alzheimer’s. Companies are already trying to treat monogenic conditions, so I think there’s a decent chance they could solve the issues of efficiently delivering gene editors to brain cells.
Problems the market might solve
Attenuating the adaptive immune response to editor proteins and delivery vectors.
If you are targeting a monogenic disease, you only need to deliver an editing vector once. The adaptive immune response to the delivery vector and CRISPR proteins is therefore of limited concern; the innate immune response to these is much more important.
There may be overlap between avoiding each of these, but I expect that if an adaptive immune response is triggered by monogenic gene therapy and it doesn’t cause anaphylaxis, companies will not concern themselves with avoiding it.Avoiding off-target edits in a multiplex editing context.
The odds of making an off-target edit scale exponentially as the number of edits increase. Rare off-targets are therefore a much smaller concern for monogenic gene therapy, and not much of a concern at all for immunotherapy unless they cause cancer or are common enough to kill off a problematic number of cells.
I therefore don’t expect companies or research labs to address this problem at the scale it needs to be addressed within the multiplex editing context.
One possible exception are companies using gene editing in the agricultural space, but the life of a wheat plant or other crop has virtually zero value, so the consequences of off-targets are far less serious.
Problems I don’t expect the market to work on
Creating better intelligence predictors.
Unless embryo selection for intelligence takes off, I think it is unlikely any of the groups that currently have the data to create strong intelligence predictors (or could easily obtain it) will do so.Conducting massive multiplex editing of human cells
If you don’t intend to target polygenic disease, there’s little motivation to make multiple genetic changes in cells. And as explained above, I don’t expect many if any people to work on polygenic disease risk because mainstream scientists are very skeptical of embryo selection and of the black box model of biology.Determining how effective edits are when conducted in adult animals
None of the monogenic diseases targeted by current therapies involve genes that are only active during childhood. It therefore seems fairly unlikely that anyone will do research into this topic since it has no clear clinical relevance.
How do we make this real?
The hardest and most important question in this post is how we make this technology real. It’s one thing to write a LessWrong post pointing out some revolutionary new technology might be possible. It’s quite another to run human trials for a potentially quite risky medical intervention that doesn’t even target a recognized disease.
Fortunately, some basic work is already being done. The first clinical trials of prime editors are likely to start in 2024. And there are at least four clinical trials underway right now testing the efficacy of base editors designed to target monogenic diseases. The VERVE-101 trial is particularly interesting due to its use of mRNA delivered via lipid nanoparticles to target a genetic mutation that causes high cholesterol and cardiovascular disease.
If this trial succeeds, it will definitively demonstrate that in-vivo gene editing with lipid nanoparticles and base editors can work. The remaining obstacles at that point will be editing efficiency and getting the nanoparticles to the brain (a solution to which I’ve proposed in the section titled “So you inject lipid nanoparticles into the brain?”. Here’s an outline of what I think needs to be done, in order:
Determine if it is possible to perform a large number of edits in cell culture with reasonable editing efficiency and low rates of off-target edits.
Run trials in mice. Try out different delivery vectors. See if you can get any of them to work on an actual animal.
Run trials in cows. We have good polygenic scores for cow traits like milk production, so we can test whether or not polygenic modifications in adult animals can change trait expression.
(Possibly in parallel with cows) run trials on chimpanzees
The goal of such trials would be to test our hypotheses about mosaicism, cancer, and the relative effect of genes in adulthood vs childhood.Run trials in humans on a polygenic brain disease. See if we can make a treatment for depression, Alzheimer’s, or another serious brain condition.
If the above passes a basic safety test (i.e. no one in the treatment group is dying or getting seriously ill), begin trials for intelligence enhancement.
It’s possible one or more steps could be skipped, but at the very least you need to run trials in some kind of model organism to refine the protocol and ensure the treatment is safe enough to begin human trials.
My current plan
I don’t have a formal background in biology. And though I learn fairly quickly and have great resources like SciHub and GPT4, I am not going to be capable of running a lab on my own any time soon.
So at the moment I am looking for a cofounder who knows how to run a basic research lab. It is obviously helpful if you have worked specifically with base or prime editors before but what I care most about is finding someone smart that I get along with who will persevere through the inevitable pains of creating a startup. If anyone reading this is interested in joining me and has such a background, send me an email.
I’m hoping to obtain funding for one year worth of research, during which our main goal will be to show we can perform a large number of distinct edits in cell culture. If we are successful in a year the odds of us being able to modify polygenic brain based traits will be dramatically increased.
At that point we would look to either raise capital from investors (if there is sufficient interest), or seek a larger amount of grant funding. I’ve outlined a brief version of additional steps as well as a rough estimate of costs below.
Short-term logistics concerns
A rough outline of my research agenda
Find a cofounder/research partner
Design custom pegRNAs for use with prime editors
Pay a company to synthesize some plasmids containing those pegRNAs with appropriate promoters incorporated
Rent out lab space. There are many orgs that do this such as BadAss labs, whose facilities I’ve toured.
Obtain other consumables not supplied by the lab space like HEK cell lines, NGS sequencing materials etc
Do some very basic validation tests to make sure we can do editing, such as inserting a fluorescent protein into an HEK cell (if the test is successful, the cell will glow)
Start trying to do prime editing. Assess the relationship between number of edits attempted and editing efficiency.
Try out a bunch of different things to increase editing efficiency and total edits.
Try multiple editing rounds. See how many total rounds we can do without killing off cells.
If the above problems can be solved, seek a larger grant and/or fundraise to test delivery vectors in mice.
Rough estimate of costs
| Cost | Purpose | Yearly Cost | Additional Notes |
| Basic Lab Space | Access to equipment, lab space, bulk discounts | ~66000 | Available from BadAss Labs. Includes workbench, our own personal CO2 incubator, cold storage, a cubicle, access to all the machines we need to do cell culturing, and the facilities to do animal testing at a later date if cell culture tests go well |
| Workers comp & liability coverage | Required by labs we rent space from | ~12,000 | Assuming workers comp is 2% of salaries, 1000 per year for general liability insurance and 3000 per year for professional liability insurance |
| Custom plasmids | gRNA synthesis | 15,000? | I’ve talked to Azenta about the pricing of these. They said they would be able to synthesize the DNA for 7000 custom guide RNAs for about $5000. We probably won’t need that many, but we’ll also likely go through a few iterations with the custom guide RNAs. |
| HEK cell lines & culture media | 2000 | Available from angioproteomie | |
| mRNA for base editors | Creating base editors in cells | 5000? | The cost per custom mRNA is around $300-400 but I assume we’ll want to try out many variants |
| mRNA for prime editors | Creating prime editors in cell cultures | 5000? | The cost per custom mRNA is around $300-400 but I assume we’ll want to try out many variants |
| Flasks or plates, media, serums, reagents for cell growth, LNP materials etc | Routine lab activity like culturing cells, making lipid nanoparticles and a host of other things | 20,000? | This is a very very rough estimate. I will have a clearer idea of these costs when I find a collaborator with whom I can plan out a precise set of experiments. |
| NGS sequencing | Determining whether the edits we tried to make actually worked | 50,000? | |
| Other things I don’t know about | 200,000? | This is a very rough estimate meant to cover lab supplies and services. I will probably be able to form a better estimate when I find a cofounder and open an account with BadAss labs (which will allow me to see their pricing for things) | |
| Travel for academic conferences, networking | I think it will likely be beneficial for one or more of us to attend a few conferences on this topic to network with potential donors. | 20,000? | |
| Salaries | Keeping us off the streets | 400,000 | There will likely be three of us working on this full time |
| Grand Total Estimate | ~795,000 |
Conclusion
Someone needs to work on massive multiplex in-vivo gene editing. This is one of the few game-board-flipping technologies that could have a significant positive impact on alignment prior to the arrival of AGI. If we completely stop AI development following some pre-AGI disaster, genetic engineering by itself could result in a future of incredible prosperity with far less suffering than exists in the world today.
I also think it is very unlikely that this will just “happen anyways” without any of us making an active effort. Base editors have been around since 2016. In that time, anyone could have made a serious proposal and started work on multiplex editing. Instead we have like two academic papers and one blog post.
In the grand scheme of things, it would not take that much money to start preliminary research. We could probably fund myself and my coauthor, a lab director, and lab supplies to do research for a year for under a million dollars. If you’re interested in funding this work and want to see a more detailed breakdown of costs than what I’ve provided above, please send me an email.
I don’t know exactly how long it will take to get a working therapy, but I’d give us a 10% chance of having something ready for human testing within five years. Maybe sooner if we get more money and things go well.
Do I think this is more important than alignment work?
No.
I continue to think the most likely way to solve alignment is for a bunch of smart people to work directly on the problem. And who knows, maybe Quintin Pope and Nora Belrose are correct and we live in an alignment by default universe where the main issue is making sure power doesn’t become too centralized in a post-AGI world.
But I think it’s going to be a lot more likely we make it to that positive future if we have smarter, more focused people around. If you can help me make this happen through your expertise or funding, please send me a DM or email me.
Appendix (Warning: boredom ahead)
Additional headaches not discussed
Regarding editing
Bystander edits
If you have a sequence like “AAAAA” and you’re trying to change it to “AAGAA”, a base editor will pretty much always edit it to “GGGGG”. This is called “bystander editing” and will be a source of future headaches. It happens because the deaminase enzyme is not precise enough to only target exactly the base pair you are trying to target; there’s a window of bases it will target. When I’ve written some software to identify which genetic variants we want to target I will be able to give a better estimate of how many edits we can target with base editors without inducing bystander edits. There are papers that have reduced the frequency of bystander edits, such as Gehrke et al, but it’s unclear to me at the moment whether the improvement is sufficient to make base editors usable on such sequences.
Chemical differences of base editor products
Traditional base editors change cytosine to uracil and adenine to inosine. Many enzymes and processes within the cell treat uracil the same as thymine and inosine the same as guanine. This is good! It’s why base editors work in the first place. But the substituted base pairs are not chemically identical.
They are less chemically stable over time.
There may be some differences in how they are treated by enzymes that affect protein synthesis or binding affinity.
It’s not clear to me at this moment what the implications of this substitution are. Given there are four clinical trials underway right now which use base editors as gene therapies, I don’t think they’re a showstopper; at most, I think they would simply reduce editing efficiency. But these chemical differences are something we will need to explore more during the research process.
Editing silenced DNA
There is mixed information online about the ability of base and prime editors to change DNA that is methylated or wrapped around histones. Some articles show that you can do this with modified base editors, but imply normal base editors have limited ability to edit such DNA. Also the ability seems to depend on the type of chromatin and the specific editor.
Avoiding off-target edits
Base editors (and to a lesser extent prime editors) have issues with off-target edits. The rate of off-targets depends on a variety of factors, but one of the most important is the similarity between the sequence of the binding site and that of other sites in the genome. If the binding site is 20 bases long, but there are several other sequences in the genome that only differ by one letter, there’s a decent chance the editor will bind to one of those other sequences instead.
But quantifying the relationship between the binding sequence and the off-target edit rate is something we still need to do for this project. We can start with models such as Deepoff, DeepCRISPR, Azimuth/Elevation, and DeepPrime to select editing targets (credit to this post for bringing these to my attention), then validate the results and possibly make adjustments after performing experiments.
Regarding fine-mapping of causal variants
There’s an annoying issue with current data on intelligence-affecting variants that reduces the odds of us editing the correct variant. As of this writing, virtually all of the studies examining the genetic roots of intelligence used SNP arrays as their data source. SNP arrays are a way of simplifying the genome sequencing process by focusing on genes that commonly vary between people. For example, the SNP array used by UK Biobank measures all genetic regions where at least ~1% of the population differs.
This means the variants that are implicated as causing an observed intelligence difference will always come from that set, even if the true causal variant is not included in the SNP array. This is annoying, because it sometimes means we’ll misidentify which gene is causing an observed difference in intelligence.
Fortunately there’s a relatively straightforward solution: in the short run we can use some of the recently released whole genome sequences (such as the 500k that were recently published by UK biobank) to adjust our estimates of any given variant causing the observed change. In the long run, the best solution is to just retrain predictors using whole genome data.
Regarding differential gene expression throughout the lifespan
Thanks to Kang et al. we have data on the level of expression of all proteins in the brain throughout the lifespan. But for reasons stated in “What if your editor targets the wrong thing”, we’re planning to make pretty much all edits to non-protein coding regions. We haven’t yet developed a mechanistic plan for linking non-coding regions with the proteins whose expression they affect. There are some cases where this should be easy, such as tying a promoter (which is always directly upstream of the exonic region) to a protein. But in other cases such as repressor genes which aren’t necessarily close to the affected gene, this may be more difficult.
Regarding immune response
I am not currently aware of any way to ensure that ONLY brain cells receive lipid nanoparticles. It’s likely they will get taken up by other cells in the body, and some of those cells will express editing proteins and receive edits. These edits probably won’t do much if we are targeting genes involved in brain-based traits, but non-brain cells do this thing called “antigen presentation” where they chop up proteins inside of themselves and present them on their surfaces.
It’s plausible this could trigger an adaptive immune response leading to the body creating antibodies to CRIPSR proteins. This wouldn’t necessarily be bad for the brain, but it could mean that subsequent dosings would lead to inflammation in non-brain regions. We could potentially treat this with an immunosupressant, but then the main advantage of lipid nanoparticles being immunologically inert kind of goes away.
Regarding brain size
Some of the genes that increase intelligence do so by enlarging the brain. Brain growth stops in adulthood, so it seems likely that any changes to genes involved in determining brain size would simply have no effect in adults. But it has been surprisingly hard to find a definitive answer to this question.
If editing genes involved in brain growth actually restarted brain growth in adulthood AFTER the fusion of growth plates in the cranium, it would cause problems.
If such a problem did occur we could potentially address it by Identifying a set of genes associated with intelligence and not brain size and exclusively edit those. We have GWAS for brain size such as this one, though we would need to significantly increase the data set size to do this.
But this is a topic we plan to study in more depth as part of our research agenda because it is a potential concern.
Editing induced amnesia?
Gene editing in the brain will change the levels and timings of various proteins. This in turn will change the way the brain learns and processes information. This is highly speculative, but maybe this somehow disrupts old memories in a way similar to amnesia.
I’m not very concerned about this because the brain is an incredibly adaptable tissue and we have hundreds of examples of people recovering functionality after literally losing large parts of their brain due to stroke or trauma. Any therapeutic editing process that doesn’t induce cancer or break critical proteins is going to be far, far less disruptive than traumatic brain injuries. But it’s possible there might be a postoperative recovery period as people’s brains adjust to their newfound abilities.
An easy way to de-risk this (in addition to animal trials) is by just doing a small number of edits during preliminary trials of intelligence enhancement.
Model assumptions
The model for gain as a function of number of edits was created by Kman with a little bit of feedback from GeneSmith.
It makes a few assumptions:
We assume there are 20,000 IQ-affecting variants with an allele frequency of >1%. This seemed like a reasonable estimation to me based on a conversation I had with an expert in the field, though there are papers that put the actual number anywhere between 10,000 and 24,000.
We use an allele frequency distribution given by the “neutral and nearly neutral mutation” model, as explained in this paper. There is evidence of current negative selection for intelligence in developed countries and evidence of past positive selection for higher-class traits (of which intelligence is almost certainly one), the model is fairly robust to effects of this magnitude.
We assume 10 million variants with minor allele frequency >1% (this comes directly from UK Biobank summary statistics, so we aren’t really assuming much here)
We assume SNP heritability of intelligence of 0.25. I think it is quite likely SNP heritability is somewhat higher, so if anything our model is conservative along this dimension
The effect size distribution is directly created using the assumption above. We assume a normal distribution of effect sizes with a mean of zero and variance set to match the estimated SNP heritability. Ideally our effect size distribution would more closely match empirical data, but this sort of effect size distribution is commonly used by other models in the field, and since it was easier to work with we used it too. But consider this an area for future improvement.
We use a simplified LD model with 100 closely correlated genetic variants. The main variable that matters here is the correlation between the MOST closely correlated SNP and the one causing the effect. We eyeballed some LD data to create a uniform distribution from which to draw the most tightly linked variant. So for example, the most tightly correlated SNP might be a random variable whose correlation is somewhere between .95 and 1. All other correlated variants are decrements of .01 less than that most correlated variant. So if the most tightly correlated variant has a correlation of .975, then the next would be correlated at .965, then .955, .945 etc for 100 variants.
This is obviously not a very realistic model, but it probably produces fairly realistic results. But again, this is an area for future improvement.
Editor properties
Here’s a table Kman and I have compiled showing a good fraction of all the base and prime editors we’ve discovered with various attributes.
| Editor | Edits that can be performed | On-target efficiency in HEK293T cells | Off-target activity in HEK293T cells | On-target edit to indel ratio | Activity window width |
| ABE7.10 | A:T to G:C | ~50% | <1% | >500 | ~3 bp |
| ABEmax | ~80% | ? | ~50 | ? | |
| BE2 | G:C to A:T | ~10% | ? | >1000 | ~5 bp |
| BE3 | ~30% | ? | ~23 | ||
| BE4max | ~60% | ? | ? | ||
| TadCBE | C:G to TA | ~57% | 0.38-1.1% | >83 | ? |
| CGBE1 | G:C to C:G | ~15% | ? | ~5 | ~3 bp |
| ATBE | A:T to T:A | ? | ? | ? | ? |
| ACBE-Q | A:T to C:G | ? | ? | ? | ? |
| PE3 | Short (~50 bp) insertions, deletions, and substitutions | ~24% | <1% | ~4.5 | N/A |
| PE3b | 41% | ~52 | |||
| PE4max | ~20% | ~80 | |||
| PE5max | ~36% | ~16 | |||
| Cas9 + HDR | Insertions, deletions, substitutions | ~0.5% | ~14% | 0.17 |
Note that these aren’t exactly apples to apples comparisons since editing outcomes vary a lot by edit site, and different sites were tested in different studies (necessarily, since not all editor variants can target the same sites)
Especially hard to report off-target rates since they’re so sequence dependent
On-target editing is also surprisingly site-dependent
Something that didn’t make the table: unintended on-target conversions for base editors
E.g. CBE converting G to C instead of G to A
Some of the papers didn’t report averages, so I eyeballed averages from the figures
If you’ve made it this far send me a DM. I want to know what motivates people to go through all of this.
- AI Alignment Metastrategy by (31 Dec 2023 12:06 UTC; 113 points)
- Four visions of Transformative AI success by (17 Jan 2024 20:45 UTC; 112 points)
- Intelligence Enhancement (Monthly Thread) 13 Oct 2023 by (13 Oct 2023 17:28 UTC; 51 points)
- 's comment on MIRI 2024 Mission and Strategy Update by (5 Jan 2024 3:48 UTC; 47 points)
- Gradient Descent on the Human Brain by (1 Apr 2024 22:39 UTC; 46 points)
- Upgrading the AI Safety Community by (16 Dec 2023 15:34 UTC; 41 points)
- 's comment on johnswentworth’s Shortform by (17 Feb 2024 6:10 UTC; 35 points)
- How to develop a photographic memory 1/3 by (28 Dec 2023 13:26 UTC; 34 points)
- 's comment on NicholasKross’s Shortform by (5 Jan 2024 3:00 UTC; 4 points)
- 's comment on MIRI 2024 Mission and Strategy Update by (6 Jan 2024 22:03 UTC; 3 points)
- 's comment on NicholasKross’s Quick takes by (EA Forum; 5 Jan 2024 3:00 UTC; 2 points)
- 's comment on D0TheMath’s Shortform by (10 Feb 2024 23:39 UTC; 2 points)
- 's comment on Significantly Enhancing Adult Intelligence With Gene Editing May Be Possible by (18 Dec 2023 0:34 UTC; 2 points)
- 's comment on ask me about technology by (29 Feb 2024 2:50 UTC; 1 point)
I am a Research Associate and Lab Manager in a CAR-T cell Research Lab (email me for credentials specifics), and I find the ideas here very interesting. I will email GeneSmith to get more details on their research, and I am happy to provide whatever resources I can to explore this possibility.
TLDR;
Making edits once your editing system is delivered is (relatively) easy. Determining which edits to make is (relatively) easy. (Though you have done a great job with your research on this, I don’t want to come across as discouraging.) Delivering gene editing mechanisms in-vivo, with any kind of scale or efficiency, is HARD.
I still think it may be possible, and I don’t want to discourage anyone from exploring this further. I think the resources and time required to bring this to anything close to clinical application will be more than you are expecting. Probably on the order of 10-20 years, at least many millions (5-10 million?) of USD, in order to get enough data to prove the concept in mice. That may sound like a lot, but I am honestly not sure if I am being appropriately pessimistic. You may be able to advance that timescale with significantly more funding, but only to a point.
Long Version:
My biggest concern is your step 1:
“Determine if it is possible to perform a large number of edits in cell culture with reasonable editing efficiency and low rates of off-target edits.”
And translating that into step 2:
“Run trials in mice. Try out different delivery vectors. See if you can get any of them to work on an actual animal.”
I would like to hear more about your research into approaching this problem, but without more information, I am concerned you may be underestimating the difficulty of successfully delivering genetic material to any significant number of cells.
My lab does research specifically on in vitro gene editing of T-cells, mostly via Lentivirus and electroporation, and I can tell you that this problem is HARD. Even in-vitro, depending on the target cell type and the amount/ it is very difficult to get transduction efficiencies higher than 70%, and that is with the help of chemicals like Polybrene, which significantly increases viral uptake and is not an option for in-vivo editing.
When we are discussing transductions, or the transfer of genetic material into a cell, efficiency measures the percentage of cells we can successfully clone a gene into.
Even when we are trying to deliver relatively small genes, we have to use a lot of tricks to get reasonable transduction efficiencies like 70% for actual human T-Cells. We Might use a very high concentration of virus, polybrene, retronectin (a protein that helps latch viruses onto a cell) and centrifuge the cells to force them into contact with the virus/retronectin.
On top of that, when we are getting transduction efficiencies of 70%, that is of the remaining cells. A significant number of the target cells will die due to the stress of viral load. I don’t know for sure how many, but I have always been told that it is typically between 30% and 70% of the cells, and the more viruses you use or, the higher transduction efficiency you go for, the more it will tend toward a higher percentage of those cells dying.
Some things to keep in mind are:
These estimates all use Lentivirus, which is considered a more efficient and less dangerous vector than AAV, mostly because it has been better studied and used.
This is all in vitro; in vivo, specialized defenses in your immune system exist to prevent the spread of viral particles. Injections of Viruses need to be localized, and you can probably only use the sort of virus that does NOT reproduce itself; otherwise, it can cause a destructive infection wherever you put it.
Your brain cannot survive 30%+ cell death. It probably can’t survive 5% cell death unless you do very small areas at a time. These transductions may have to happen for every gene edit you want to make based solely on currently available technology.
Mosaicism is probably not a problem, but keep in mind that there is a selective effect since cases where it is a problem are selected out of your observations since if they are destructive, they won’t be around to be observed. This, of course, would be easily tested out.
Essentially, in order to make this work for in-vivo gene editing of an entire organ (particularly the brain), you need your transduction efficiency to be at least 2-3 orders of magnitude higher than the current technologies allow on their own just to make up for the lack of polybrene/retronectin in order to hit your target 50%. The difference in using Polybrene and Retronectin/Lenti-boost is the difference between 60% transduction efficiency and 1%. You may be able to find non-toxic alternatives to polybrene, but this is not an easy problem, and if you do find something like that, it is worth a ton of money and/or publication credit on its own.
I don’t want to be discouraging here; however, it is important to understand the problem’s real scope.
At a glance, I would say the adenoviral approach is the most likely to work for the scale of transduction you are looking to accomplish. After a quick search, I found these two studies to be the most promising, discussing the deployment of CRISPR/Cas9 systems via AAV. Both use hygromycin B selection (a process whereby cells that were not transduced are selected out since hygromycin will kill the cells that don’t have the immunity sequence included in the Cas9 package.) and don’t mention specific transduction efficiency numbers, but I am guessing it is not on the order of 50%. At most I would hope it is as high as 5%.
All of this does not account for the other difficulties of passive immunity of gene editing in vivo.
Why aren’t others doing this?
I think I can help answer this question. The short answer is that they are, but they are doing it in much smaller steps. Rather than going straight for the holy grail of editing an organ as large and complex as the brain, they are starting with cell types and organs that are much easier to make edits to.
This Paper is the most recent publication I can find on in-vivo gene editing, and it discusses many of the strategies you have highlighted here. In this case, they are using Lipid Nano-Particles to target specifically the epithelial cells of the lungs to edit out the genetic defect that causes Cystic Fibrosis. This is a much smaller and more attainable step for a lot of reasons, the biggest one being that they only need to attain a very low transduction efficiency to have a highly measurable impact on the health of the mice they were testing on. It is also fairly acceptable to have a relatively high rate of cell death in epithelial cells since they replace themselves very rapidly. In this case, their highest transduction efficiency was estimated to be as high as 2.34% in-vivo, with a sample size of 8 mice.
We may be able to quickly come up with at least one meaningful gene target that could make a difference with 2.34% transduction efficiency, but be aware that delivering this at scale to a human brain will be MUCH harder than doing so with mouse epithelial cells.
Again, I don’t want to discourage this project. I would really like to help, actually. I want to be realistic about the challenges here, and there is a reason why the equilibria is where it is.
Thanks for leaving such a high quality comment. I’m sorry for taking so long to get back to you.
We fully expect bringing this to market to take tens of millions of dollars. My best guess was $20-$40 million.
We expect this to be difficult, but we DON’T expect to have to solve the delivery problem entirely on our own. There are significant incentives for existing companies such as Dyno Therapeutics to solve the problem of delivering genes (or other payloads) to the nucleus of brain cells. In fact, Dyno already has a product, Dyno bCap 1 which successfully delivered genes to between 5% and 20% of brain cells in non-human primates.
Obviously we will need higher efficiencies than that to perform edits for polygenic brain diseases or intelligence, but the ease of delivering payloads to brain cells has been gradually improving over the years and I expect it to continue doing so.
There are of course some issues:
I know from some conversations with a former employee of Dyno that the capsids can be customized to be serologically distinct so that any antibodies formed in response to one round of treatment will not destroy the capsids used in the second round. But I am still waiting to hear back from them regarding the cost and time required to do this sort of customization.
Custom AAVs are also quite expensive per dose, largely due to the cost of reagents and other basic supplies that no one has figured out how to make cheaper yet. So it’s a plausible delivery mechanism, but far from ideal.
Still, the fact that there is an existing product on the marketplace which can get custom DNA payloads into the nuclei of brain cells gives me hope that someone else will have made major headway on the delivery problem by the time we are ready for trials in cows or non-human primates.
In the meantime, we simply need a way to get editors into HEK cells and brain cells efficiently in cell cultures to test multiplex editing approaches. I’m sure this will pose its own set of challenges, but given dozens of labs have done this I don’t expect that to be infeasible.
Send me an email if you have time to chat about this. If you’re willing I’d like to pick your brain more about other aspects of the project.
Really interesting, thanks for commenting.
Are you doing traditional gene therapy or CRISPR-based editing?
If the former, I’d guess you’re using Lentivirus because you want genome integration?
If the latter, why not use Lipofectamine?
How do you use electroporation?
Does this refer to the proportion of the remaining cells which had successful edits / integration of donor gene? Or the number that were transfected at all (in which case how is that measured)?
This study achieved up to 59% base editing efficiency in mouse cortical tissue, while this one achieved up to 42% prime editing efficiency (both using a dual AAV vector). These contributed to our initial optimism that the delivery problem wasn’t completely out of reach. I’m curious what you think of these results, maybe there’s some weird caveat I’m not understanding.
This is my belief as well—though the dearth of results on multiplex editing in the literature is strange. E.g. why has no one tried making 100 simultaneous edits at different target sequences? Maybe it’s obvious to the experts that the efficiency would be to low to bother with?
I’ll give a quick TL;DR here since I know the post is long.
There’s about 20,000 genes that affect intelligence. We can identify maybe 500 of them right now. With more data (which we could get from government biobanks or consumer genomics companies), we could identify far more.
If you could edit a significant number of iq-decreasing genetic variants to their iq-increasing counterpart, it would have a large impact on intelligence. We know this to be the case for embryos, but it is also probably the case (to a lesser extent) for adults.
So the idea is you inject trillions of these editing proteins into the bloodstream, encapsulated in a delivery capsule like a lipid nanoparticle or adeno-associated virus, they make their way into the brain, then the brain cells, and the make a large number of edits in each one.
This might sound impossible, but in fact we’ve done something a bit like this in mice already. In this paper, the authors used an adenovirus to deliver an editor to the brain. They were able to make the targeted edit in about 60% of the neurons in the mouse’s brain.
There are two gene editing tools created in the last 7 years which are very good candidates for our task, with a low chance of resulting in off-target edits or other errors. Those two tools are called base editors and prime editors. Both are based on CRISPR.
If you could do this, and give the average brain cell 50% of the desired edits, you could probably increase IQ by somewhere between 20 and 100 points.
What makes this difficult
There are two tricky parts of this proposal: getting high editing efficiency, and getting the editors into the brain.
The first (editing efficiency) is what I plan to focus on if I can get a grant. The main issue is getting enough editors inside the cell and ensuring that they have high efficiency at relatively low doses. You can only put so many proteins inside a cell before it starts hurting the cell, so we have to make a large number of edits (at least a few hundred) with a fixed number of editor proteins.
The second challenge (delivery efficiency) is being worked on by several companies right now because they are trying to make effective therapies for monogenic brain diseases. If you plan to go through the bloodstream (likely the best approach), the three best candidates are lipid nanoparticles, engineered virus-like particles and adeno-associated viruses.
There are additional considerations like how to prevent a dangerous immune response, how to avoid off-target edits, how to ensure the gene we’re targeting is actually the right one, how to get this past the regulators, how to make sure the genes we target actually do something in adult brains, and others which I address in the post.
What I plan to do
I’m trying to get a grant to do research on multiplex editing. If I can we will try to increase the number of edits that can be done at the same time in cell culture while minimizing off-targets, cytotoxicity, immune response, and other side-effects.
If that works, I’ll probably try to start a company to treat polygenic brain disorders like Alzheimers. If we make it through safety trials for such a condition, we can probably start a trial for intelligence enhancement.
If you know someone that might be interested in funding this work, or a biologist with CRISPR editor expertise, please send me a message!
Why would this work on adults? The brain develops most in childhood. If those genes’ role is to alter the way synapses develop in the fastest growth phase, changing them when you’re 30 won’t do anything.
The hope is that local neural function could be altered in a way that improves fluid intelligence, and/or that larger scale structural changes could happen in response to the edits (possibly contingent on inducing a childlike state of increased plasticity).
The former thing sounds like overclocking a CPU. The latter instead “erase chunks of someone’s personality and memory and let them rewrite it, turning them into an essentially different person”. I don’t think many people would just volunteer for something like that. We understand still far too little of how brains work to think that tinkering with genes and just getting some kind of Flowers for Algernon-ish intelligence boost is the correct model of this. As it often happens, it’s much easier to break something than to build it up, especially something as delicate and complex as a human brain. Right now this seems honestly to belong in the “mad science” bin to me.
This reply is hilarious in the context of your first one. At first you confidently assert that changing genes in the brain won’t do anything to an adult, followed by your statement that “we understand still far too little of how brains work” to know what’s going to happen following such a therapy along with other predictions like total memory erasure. Which is it?
While the vast majority of neurons are subject to mitotic arrest after adolescence, gene expression, the regulation of gene expression, and morphological/biochemical restructuring of individual neurons (plasticity) doesn’t stop until you’re dead. Additionally, there’s no reason to rule out the possibility of genome changes in adolescence leading to macroscopic changes in brain structure, especially considering that certain histone de-acetylase inhibitors like valproate have been shown to re-activate developmental critical periods in particular areas of the brain, and that cell reprogramming therapies such as those being interrogated by David Sinclair’s lab have been demonstrated to be able to rewind the biological clock on neurons, regaining any lost plasticity, regeneration ability, or otherwise anti-change properties gained with age. Even without such fancy therapies, there is nothing barring the possibility that macroscopic brain morphology is to an extent emergent from gene expression at the level of individual neurons, and that changes at the local scale could reverberate globally to generate different macroscopic morphological characteristics.
But suppose that all of what I said isn’t true, and indeed genetic changes as an adolescent would not lead to the macroscopic morphological characteristics that have been shown to correlate with intelligence, like greater brain volume, shorter white matter path lengths, greater cortical thickness, etc. To kman’s point, alterations at the level of local neurons can still be beneficial for intelligence, which has already been demonstrated unlike my hypotheticals in the last paragraph. This is obviously true to anyone who performs a quick survey of some of the SNPs associated with greater intelligence, such as having a T allele at rs2490272, which is on the FOXO3 gene. The protein encoded by FOXO3 isn’t involved in the macrosopic structural formation of the brain whatsoever, and instead acts on the cellular level to protect neurons from oxidative stress, regulate protein turnover, and regulate DNA repair. It is easy to see why having a better version of this protein would lead to healthier neurons (whatever that means) and thus greater intelligence, which is what all GWASes that investigate this gene show. FOXO3 is just one of many of such genes.
I mean, you sound like you know far more than me on it so I won’t argue the specifics, but in general, “we know enough about this thing to not be able to safely mess with it, but to be reasonably sure that messing with it will have bad effects” is absolutely possible. It’s in fact the default for really complex black boxes: while understanding their general function may be easy enough, if you don’t know what you’re doing messing with their internals, odds are that you’ll break them rather than improve them.
The prediction of “total memory erasure” was a response to a specific idea, the notion that if intelligence was really mostly determined in childhood/adolescence, then if you could push the brain to regain its original plasticity you could repeat the process with different outcomes, and as I said that sounds like it would change a lot about what a person is (unless you can somehow disentangle experiences and personality from intelligence gained through them). I don’t expect that to be the case if one of the premises doesn’t hold; I was just criticizing that specific strategy. Others would not have this downside.
As for the rest, sure, it’s possible that there might be gene changes that will simply improve neuron health. But if I had to bet I’d imagine it would be easier to gain traits like resistance to dementia and Alzheimer’s or such by tweaking those, than a whole 40 points of IQ or such. I know brains aren’t the same as ANNs, but to make an analogy, if you run GPT-4 on newer hardware it’ll do the same things a bit better and faster, but it won’t be able to make entirely new things out of whole cloth.
Yes, as someone who has worked both in CS and in neuroscience at the graduate level, I probably do know far more than you about this topic. At the risk of sounding more polemic than I am, it’s posts like yours and others that make excessively reductive inferences about neurons and the brain that invariably end up polluting discussions of this topic and turn it into an unproductive cesspool of ML people offering their two-cents for topics they don’t understand (most of the replies to the original post being the cases in point).
I will grant you that it is indeed possible that we don’t understand enough about the brain to be confident that we won’t just irreversibly ruin test subjects’ brains with such a therapy, but this is much less likely than the possibility that either nothing will happen or that gains will be had, provided such a therapy is successfully developed in a way that makes it feasible. Geneticists and neuroscientists have not been doing nothing for the past century; we understand much more about the brain, neurons, and cell biology than we understand about artificial neural networks, which is why we can be confident that things like this are possible if we can overcome the obstacles in engineering the delivery and editing mechanisms. There is also no reason to get it right on the first try; GeneSmith has sadly had to state the obvious several times in response to others, which is that we have the scientific method and it would take many experiments to get such a therapy right if it is at all possible. Regardless, I don’t think this therapy as OP describes it is possible for reasons that have already been stated by HiddenPrior and other reasons, but not for the inane reasons others have suggested that liken the adult brain to an immutable pre-trained neural net. It will take several breakthroughs in the field before reliable multiplex genome editing in the human central nervous system becomes a reality.
You are right however that without more GWASes, it will likely be impossible to extricate intelligence enhancements and changes to things like one’s psychology and predisposition to psychiatric disorders. It is even possible that these end up being inextricable to an extent, and recipients of this therapy would have to accept sustaining some degree of not only personality change but other introspective “updates” and disease risk changes. This is one aspect of this therapy that the OP has been rather naïve about in the post and replies to others. If a gene’s influence is such that it affects as emergent and complex a trait as intelligence, it is reasonable to suspect that it affects other things. This is demonstrable with GWASes (and has been demonstrated), but the silver lining is that allele flips that enhance intelligence tend to confer positive rather than negative changes to other systems (key word here being “tend”). As far as my personal preference goes, I would gladly accept a different personality if it meant having an IQ of 190+ or something; nonetheless, there’s no reason to believe personality isn’t amenable to the same techniques used to alter intelligence.
The bit about the personality was specifically in response to the idea that you could revert brains to childhood-like plasticity. That’s like an additional layer of complexity and unlike gene therapy we don’t know how to begin doing that, so if you ask me, I don’t think it would actually be a thing anyway in the near future. My guess is: most of your intelligence, even the genetic component, is probably determined by development during the phase of highest plasticity. So if you change the genes later you’ll either get no effect or marginal ones compared to what would happen if you changed them in embryos—that is, if it doesn’t also cause other weird side effects.
Experiments are possible but I doubt they’d be risk-free, or honestly, even approved by an ethical committee at all, as things are now. It’s a high risk for a goal that would probably be deemed in itself ethically questionable. And the study surviving for example a cohort “gone bad” would be really hard in terms of public support and funding.
Can you elaborate on this? We’d really appreciate the feedback.
I posted my reply to this as a direct reply to the OP because I think it’s too huge and elaborate to keep hidden here.
I just want to point out that the sentence you replied to starts with an “if”. “If those genes’ role is to alter the way synapses develop in the fastest growth phase, changing them when you’re 30 won’t do anything” (emphasis mine). You described this as “At first you confidently assert that changing genes in the brain won’t do anything to an adult”. The difference is important. This is in no way a comment on the object level debate. I simply think Lesswrong is a place where hypotheticals are useful and that debates will be poorer if people cannot rely on the safety that saying “if A then B” will not be interpreted as just saying “B”.
I agree, it seems totally bizarre to imagine even a single person reading this post would genuinely want to erase themselves in order to make room for a ‘smarter’ personality.
Even if it was offered for free with no other risks or side effects whatsoever. Let alone in the real world.
I’m not sure if this objection has been pointed out or is even valid.. I think the argument from approximate linearity is probably wrong, even if we’re talking editing embryos and not adults. In machine learning we make the learning rate small enough that the map of the error over the parameter space appears linear. This means scaling the gradients way down, but my intuition is that it’s minimizing the euclidean distance covered by each step that’s “doing the work” of making everything appear flat. If that’s correct then flipping 20000 genes is a massive step through gene space compared to flipping just a few and linearity would likely break down. I would expect you can beat sexual selection with methods like you describe, since we can use population studies to get a nice accurate estimate of that “gradient”, but getting to IQ 900 or whatever seems a stretch to put it mildly.
I mean, I explicitly state in the post that I don’t think we’ll be able to reach IQs far outside the normal human range by just flipping alleles:
So yes, I agree with you
I have to confess that I did some skimming, and by ctrl-f it looks like I actually read right up to the first half of that paragraph before I got lazy. Fwiw it was due to mental and time constraints and nothing to do with the quality of writing.
ANNs and BNNs operate on the same core principles; the scaling laws apply to both and IQ in either is a mostly function of net effective training compute and data quality. Genes determine a brain’s architectural prior just as a small amount of python code determines an ANN’s architectural prior, but the capabilities come only from scaling with compute and data (quantity and quality).
So you absolutely can not take datasets of gene-IQ correlations and assume those correlations would somehow transfer to gene interventions on adults (post training in DL lingo). The genetic contribution to IQ is almost all developmental/training factors (architectural prior, learning algorithm hyper params, value/attention function tweaks, etc) which snowball during training. Unfortunately developmental windows close and learning rates slow down as the brain literally carves/prunes out its structure, so to the extent this could work at all, it is mostly limited to interventions on children and younger adults who still have significant learning rate reserves.
But it ultimately doesn’t matter, because the brain just learns too slowly. We are now soon past the point at which human learning matters much.
How do you know this?
In comparing human brains to DL, training seems more analogous to natural selection than to brain development. Much simpler “architectural prior”, vastly more compute and data.
We’re really uncertain about how much would transfer! It would probably affect some aspects of intelligence more than others, and I’m afraid it might just not work at all if g is determined by the shape of structures that are ~fixed in adults (e.g. long range white matter connectome). But it’s plausible to me that the more plastic local structures and the properties of individual neurons matter a lot for at least some aspects of intelligence (e.g. see this).
There’s a lot more to intelligence than learning. Combinatorial search, unrolling the consequences of your beliefs, noticing things, forming new abstractions. One might consider forming new abstractions as an important part of learning, which it is, but it seems possible to come up with new abstractions ‘on the spot’ in a way that doesn’t obviously depend on plasticity that much; plasticity would more determine whether the new ideas ‘stick’. I’m bottlenecked by the ability to find new abstractions that usefully simplify reality, not having them stick when I find them.
My model is there’s this thing lurking in the distance, I’m not sure how far out: dangerously capable AI (call it DCAI). If our current civilization manages to cough up one of those, we’re all dead, essentially by definition (if DCAI doesn’t kill everyone, it’s because technical alignment was solved, which our current civilization looks very unlikely to accomplish). We look to be on a trajectory to cough one of those up, but It isn’t at all obvious to me that it’s just around the corner: so stuff like this seems worth trying, since humans qualitatively smarter than any current humans might have a shot at thinking of a way out that we didn’t think of (or just having the mental horsepower to quickly get working something we have thought of, e.g. getting mind uploading working).
From study of DL and neuroscience of course. I’ve also written on this for LW in some reasonably well known posts: starting with The Brain as a Universal Learning Machine, and continuing in Brain Efficiency, and AI Timelines specifically see the Cultural Scaling Criticality section on the source of human intelligence, or the DL section of simboxes. Or you could see Steven Byrne’s extensive LW writings on the brain—we are mostly in agreement on the current consensus from computational/systems neuroscience.
The scaling laws are extremely well established in DL and there are strong theoretical reasons (and increasingly experimental neurosci evidence) that they are universal to all NNs, and we have good theoretical models of why they arise. Strong performance arises from search (bayesian inference) over a large circuit space. Strong general performance is strong performance on many many diverse subtasks which require many many specific circuits built on top of compressed/shared base circuits down a heirarchy. The strongest quantitative predictor of performance is the volume of search space explored which is the product of C * T (capacity and data/time). Data quality matters in the sense that the search volume quantitative function of predictive loss only applies to tasks similar enough to the training data distribution.
No—biological evolution via natural selection is very similar to technological evolution via engineering. Both brains and DL systems have fairly simple architectural priors in comparison to the emergent learned complexity (remember whenever I use the term learning, I use it in a technical sense, not a colloquial sense) - see my first early ULM post for a review of the extensive evidence (greatly substantiated now by my scaling hypothesis predictions coming true with the scaling of transformers which are similar to the archs I discussed in that post).
Whenever I use the word learning, without further clarification, I mean learning as in bayesian learning or deep learning, not in the colloquial sense. My definition/sense of learning encompasses all significant changes to synapses/weights and is all encompassing.
Brains are very slow so have limited combinatorial search, and our search/planning is just short term learning (short/medium term plasticity). Again it’s nearly all learning (synaptic updates).
I find the standard arguments for doom implausible—they rely on many assumptions contradicted by deep knowledge of computational neuroscience and DL.
I was at the WBE2 workshop with Davidad but haven’t yet had time to write about progress (or lack thereof); I think we probably mostly agree that the type of uploading moonshot he discusses there is enormously expensive (not just in initial R&D, but also in recurring per scan costs). I am actually more optimistic than more pure DL based approaches will scale to much lower cost, but “much lower cost” is still on order of GPT4 training cost just to process the scan data through a simple vision ANN—for a single upload.
I’m not aware of these—do you have any references?
True but misleading? Isn’t the brain’s “architectural prior” a heckuva lot more complex than the things used in DL?
Sure. The big crux here is whether plasticity of stuff which is normally “locked down” in adulthood is needed to significantly increase “fluid intelligence” (by which I mean, something like, whatever allows people to invent useful new concepts and find ingenious applications of existing concepts). I’m not convinced these DL analogies are useful—what properties do brains and deepnets share that renders the analogies useful here? DL is a pretty specific thing, so by default I’d strongly expect brains to differ in important ways. E.g. what if the structures whose shapes determine the strength of fluid intelligence aren’t actually “locked down”, but reach a genetically-influenced equilibrium by adulthood, and changing the genes changes the equilibrium? E.g. what if working memory capacity is limited by the noisiness of neural transmission, and we can reduce the noisiness through gene edits?
FOOM isn’t necessary for doom—the convergent endpoint is that you have dangerously capable minds around: minds which can think much faster and figure out things we can’t. FOOM is one way to get there.
[Scaling law theories]
Sure: here’s a few: quantization model, scaling laws from the data manifold, and a statistical model.
The full specification of the DL system includes the microde, OS, etc. Likewise much of the brain complexity is in the smaller ‘oldbrain’ structures that are the equivalent of a base robot OS. The architectural prior I speak of is the complexity on top of that, which separates us from some ancient earlier vertebrate brain. But again see the brain as a ULM post, which cover the the extensive evidence for emergent learned complexity from simple architecture/algorithms (now the dominant hypothesis in neuroscience).
Most everything above the hardware substrate—but i’ve already provided links to sections of my articles addressing the convergence of DL and neurosci with many dozens of references. So it’d probably be better to focus exactly on what specific key analogies/properties you believe diverge.
DL is extremely general—it’s just efficient approximate bayesian inference over circuit spaces. It doesn’t imply any specific architecture, and doesn’t even strongly imply any specific approx inference/learning algorithm (as 1st and approx 2nd order methods are both common).
Training to increase working memory capacity has near zero effect on IQ or downstream intellectual capabilities—see gwern’s reviews and experiments. Working memory capacity is important in both brains and ANNs (transformers), but it comes from large fast weight synaptic capacity, not simple hacks.
Noise is important for sampling—adequate noise is a feature, not a bug.
So I agree with your general point that genetic interventions made in adults would have a lesser effect than those same interventions made in embryos, which is why our model assumes that the average genetic change would have just half the normal effect. The exact relative size of edits made in the adult brain vs an embryo IS one of the major unknown factors in this project but like… if brain size were the only thing affecting intelligence we’d expect a near perfect correlation between it and intelligence. But that’s not what we see.
Brain size only correlates with intelligence at 0.3-0.4.
So there’s obviously a lot more going on.
It’s not post-training. Brains are constantly evolving and adapting throughout the lifespan.
If this was actually the case then none of the stuff people are doing in AI safety or anything else would matter. That’s clearly not true.
We can roughly bin brain tissue into 3 developmental states:
juvenile: macro structure formation—brain expanding, neural tissue morphogenesis, migration, etc
maturing: micro synaptic structure formation, irreversible pruning and myelination
mature: fully myelinated, limited remaining plasticity
Maturation proceeds inside out with the regions closest to the world (lower sensory/motor) maturing first, proceeding up the processing hierarchy, and ending with maturation of the highest levels (some prefrontal, associative etc) around age ~20.
The human brain’s most prized intellectual capabilities are constrained (but not fully determined) mostly by the upper brain regions. Having larger V1 synaptic capacity may make for a better fighter pilot through greater visual acuity, but STEM capability is mostly determined by capacity & efficiency of upper brain regions (prefrontal, associative, etc and their cerebellar partners).
I say constrains rather than determines because training data quantity/quality also obviously constrains. Genius level STEM capability requires not only winning the genetic lottery, but also winning the training run lottery.
IQ itself only correlates with STEM potential (and less so as you move away from the mean) but sure there are many ways to make a brain larger that do not specifically increase synaptic capacity&efficiency of the specific brain regions most important for STEM capability. Making neurons larger, increasing the space between them, increasing glial size or counts, etc. But some brain size increase methods will increase the size of STEM linked brain regions, so 0.3-0.4 seems about right.
The capacity&efficiency of the most important brain regions is mostly determined by genes effecting the earliest stage 1 - the architectural prior. These regions won’t fully be used until ~20 years later due to how the brain trains/matures modules over time, but most of the genetic influence is in stage 1 - i’d guess 75%.
I’d guess the next 20% of genetic influence is on stage 2 factors that effect synaptic efficiency and learning efficiency, and only 5% influence on 3 via fully mature/trained modules.
Yes a few brain regions (hippocampus, BG, etc) need to maintain high plasticity (with some neurogenesis) even well in to adulthood—they never fully mature to stage 3. But that is the exception, not the rule.
Not really—See above. At 45 most of my brain potential is now fully spent. I’m very unlikely to ever be a highly successful chess player or physicist or poet etc. Even learning a new human language is very slow and ineffective compared to a child. It’s all about depletion of synaptic learning potential reserves.
The colloquial use of the word ‘learning’ as in ‘learning’ new factual information is not at all what I mean and is not relevant to STEM capability. I am using ‘learning’ in the more deep learning sense of learning deep complex circuits important for algorithmic creativity, etc.
As a concrete specific example, most humans learn to multiply large numbers by memorizing lookup tables for multiplication of individual digits and memorizing a slow serial mental program built on that. But that isn’t the only way! It is possible to learn more complex circuits which actually do larger sum addition&multiplication directly—and some mentats/savants do acquire these circuits (with JVN being a famous likely example).
STEM capability is determined by deep learning many such circuits, not ‘learning’ factual knowledge.
Now it is likely that one of the key factors for high intelligence is a slower and more efficient maturation cycle that maintains greater synaptic learning reserves far into adulthood—ala enhanced neotany, but that is also an example of genetic influence that only matters in stage 1 and 2. Maturation is largely irreversible—once most connections are pruned and the few survivors are strengthened/myelinated you can’t go back to the earlier immature state of high potential.
Huh? Oh—by learning there I meant full learning in the training sense—stages 1 and 2. Of course things adults do now matter, they just don’t matter through the process of training new improved human brains.
Couldn’t there be genetic effects on things that can improve the brain even once its NN structure is mostly fixed? Maybe it’s possible to have neurons work faster, or for the brain to wear less with abstract thinking, or to need less sleep.
This kind of thing is not a full intelligence improvement because it does not allow you to notice more patterns or to think with new schemes.
So maybe yes, it won’t make a difference for AI timelines, though it would still be a very big deal.
Sure—I’m not saying no improvement is possible. I expect that the enhancements from adult gene editing should encompass most all of the brain tweaks you can get from drugs/diet. But those interventions will not convert an average brain into an Einstein.
The brain—or more specifically the brains of very intelligent people—are already very efficient, so I’m also just skeptical in general that there are many remaining small tweaks that take you past the current “very intelligent”. Biological brains beyond the human limit are of course possible, but probably require further significant size expansion amongst other infeasible changes.
Sleep is very important, less isn’t really better—most of the critical cortex learning/training happens during sleep through episodic replay, SWRs and REM etc.
See Would edits to the adult brain even do anything?.
(Not endorsing the post or that section, just noticing that it seems relevant to your complaint.)
It does not. Despite the title of that section it is focused on adult expression factors. The post in general lacks a realistic mechanistic model of how tweaking genes affects intelligence.
Is similar to expecting that a tweak to the hyperparams (learning rate) etc of trained GPT4 can boost its IQ (yes LLMs have their IQ or g factor). Most all variables that affect adult/trained performance do so only through changing the learning trajectory. The low hanging fruit or free energy in hyperparams with immediate effect is insignificant.
Of course if you combine gene edits with other interventions to rejuvenate older brains or otherwise restore youthful learning rate more is probably possible, but again it doesn’t really matter much as this all takes far too long. Brains are too slow.
We thought a bit about this, though it didn’t make the post. Agree that it increases the chance of the editing having a big effect.
Maybe it’s the lack of sleep for me but is “Brains are too slow.” referring to something like growth/formation of structures that support some level of intelligence or to to human brain’s just being slower than and AGI?
Too slow too matter now due to the slow speed of neurons and bio learning combined with where we are in AI.
Why think the brain learns too slowly? If I can boost my sample efficiency I can learn new subjects quicker, remember more facts, and do better thought-result attribution. All these seem short-term beneficial. Unless you think there’s major critical period barriers here, these all seem likely results.
Though I do agree that a person with the genes of a genius for 2 years will be far less a genius than a person with the genes of a genius for 25 years. It seems a stretch to say the first change can be rounded off as not matteringthough.
It would matter in a world without AI, but that is not the world we live in. Yes if you condition on some indefinite AI pause or something then perhaps, but that seems extremely unlikely. It takes about 30 years to train a new brain—so the next generation of humans won’t reach their prime until around the singularity, long after AGI.
Most genius is determined prenatally and during ‘training’ when cortex/cerebellum modules irreversibly mature, just as the capabilities of GPT4 are determined by the initial code and the training run.
I think I agree with everything that you said except that it won’t matter. It seems like it’d very much matter if in the most successful case we make people learn skills, facts, and new fields 6x faster. Maybe you think 6x is too high, and its more like 1.05x, enough to notice a difference over 30 years, but not over 2-5.
So other than the medical issues which makes this idea unviable (off target edits causing cancer, major brain firmware edits causing uncontrollable seizures), we can also bound our possible performance increases.
We haven’t increased brain volume meaningfully, the patients skull plates are fixed. And they still have one set of eyes and one set of hands. Nerve transmission velocities haven’t been improved either.
In terms of real performance is 6x achievable? Does it mean anything? Even the most difficult tasks humans have ever accomplished require taking in the latest data from the last round of testing and then choosing what to try next based on this information. This “innovation feedback cycle” is several steps, and the “think about it” step, even if we posit we can make it infinitely fast, would be limited by “view the data” and “communicate with other people” steps.
That is I am taking a toy model:
View the data, think about the next experiment, tell someone/something else to do the experiment
Are the only 3 steps.
If the view/tell steps take more than 1⁄6 of the total time 6x performance increase is impossible. This is Amdahls law.
Viewing and telling are themselves learned skills that can be sped up. Most are far from their maximal physically possible reading/listening speed, or their maximal physically possible writing speed. For example, just after high school I tried to read sutton & barto and spent a week reading each chapter. Later I read it & spent a day on each chapter. That’s a 7x improvement just from meta-learning!
You’re still i/o limited though. And optimizations you mention come with tradeoffs, skipping words by speed reading for example won’t work at all when the content has high information density
I still read every word, I just knew better what to think about, recall was faster, etc. I was reading at a leisurely pace as well. If you want to call learning what to pay attention to & how to pay attention to it not an i/o problem, just the physical limits, then I do think i/o is very very fast, taking <<1/6 of time.
Depends on what it is. Experimenting with AI? Fixing cars? Working on particle physics as CERN? Developing the atomic bomb? Discovering the mass of the electron? Performing surgery? Developing new surgery methods?
I/O is at least 90 percent of each of those tasks.
I don’t know how to think about i/o in the tasks you mention, so I don’t think the question is very useful. Definitely on an individual level, much time is spent on i/o, but that’s beside the point, as I said above people can do more efficient i/o than they currently do, and generally smarter people are able to do more efficient i/o. When I ask myself why we aren’t better at the tasks you mention, mostly I think firstly we are coordination constrained, and secondly we are intelligence constrained. Maybe we’re also interface constrained, which seems related to i/o, but generally we make advancements in interfaces, and this improves productivity in all the tasks you mention, and smarter people can use & make smarter interfaces if that is indeed the problem.
A good motivator: There exist 10x software engineers, who are generally regarded as being 10x better programmers than regular engineers. If i/o was the limiter for programming ability, then such people would be expected to simply have better keyboards, finger dexterity, and eyesight. Possibly they have these to some extent, but I expect their main edge over other 1x engineers is greater sample efficiency when generalizing from one programming task to another. We can thus conclude that i/o takes up <1/10 the time in programming. Probably <<1/10.
There also probably exist 10x surgeons, experimentalists, and mechanics. Perhaps there also exist 10x particle physicists at CERN, though there are fewer of them, and it may be less obvious.
So if 10x software engineers exist, they develop architecture and interfaces and use patterns where over time 1⁄10 the total amount of human time per feature is used. Bad code consumes enormous amounts of time to deal with, where bad architecture that blocks adding new features or makes localizing a bug difficult would be the worst.
But to be this good mostly comes from knowledge, learned either in school or over a lot of time from doing it the wrong way and learning how it fails.
It’s not an intelligence thing. A genius swe can locate a bug on a hunch, a 10x swe would write code where the bug doesn’t exist or is obvious every time.
A lot of the other examples I gave I have the impression that no, I/o is everything. Finding the mass of the electron was done with laborious effort over many hours, most of it dealing with the equipment. Nobody can cut 10 times faster in surgery, hands can’t run that quickly. Same with fixing a car. Cern scientists obviously are limited by all sorts of equipment issues. Same with AI research—the limiting factor has always been equipment from the start. “Equipment issues” mean either you get your hands dirty fixing it yourself—that’s I/O or spare parts bound—or you tell someone else to do it and their time to fix is bound the same way.
Some of the best scientists in history could fix equipment issues themselves, this likely broadened their skill base and made their later discoveries feasible.
You are operating on the wrong level of analysis here. The question is about skill improvement, not execution.
They aren’t the same thing? I mean for the topics of interest, AI alignment, there is nothing to learn from other humans or improve on past a certain baseline level of knowledge. Past a certain point reading papers on it I suspect your learning curve would go negative because you’re just learning on errors people before you made.
Improving past that point has to be designing and executing high knowledge gain experiments, and that’s I/o and funding bound.
I would argue that the above is the rule for anything humans cannot already do.
Were you thinking of skills where it’s a confined objective task? Like StarCraft 2 or Go? The former being strongly I/o bound.
I’m very confident we’re talking past each other, and I’m not in the mood to figure out what we actually disagree on. I think we’re using “i/o” differently, and I claim your use permits improvements to the process, which contradicts your argument.
I am not so sure about that. I am thinking back to the Minnesota Twin Study here, and the related fact that heritability of IQ increases with age (up until age 20, at least). Now, it might be that we’re just not great at measuring childhood IQ, or that childhood IQ and adult IQ are two subtly different things.
But it certainly looks as if there’s factors related to adult brain plasticity, motivation (curiosity, love of reading, something) that continue to affect IQ development at least until the age of 18.
Straight forward result of how the brain learns. Cortical/cerebellar modules start out empty and mature inwards out—starting with the lowest sensory/motor levels closest to the world and proceeding up the hierarchy ending with the highest/deepest modules like prefrontal and associative cortex. Maturation is physically irreversible as it involves pruning most long-range connections and myelinating&strengthening the select few survivors. Your intelligence potential is constrained prenatally by genes influencing synaptic density/connectivity/efficiency in these higher regions, but those higher regions aren’t (mostly) finishing training until ~20 years age.
Is it true? We need to pour lifetimes of information to get moderate expertize-level performance in SOTA models. I have no significant doubt that we can overcome this via scaling, but with correction on available compute, brains seem to be decent learners.
In addition, I would say that here is a difference between learning capability and elicting it: current models seem to be very sensitive to prompts, wrappnigs and other conditions. It’s possible that intelligence gains can come from easier eliciting of already learned capabilities but blocked by, say, social RLHF.
Current AI is less sample efficient, but that is mostly irrelevant as the effective speed is 1000x to 10000x greater.
By the time current human infants finish ~30 year biological training, we’ll by long past AGI and approaching singularity (in hyperexpoential models).
Regulation and complexity of effects seem like another two big blockers.
Effects of genes are complex. Knowing a gene is involved in intelligence doesn’t tell us what it does and what other effects it has.
I wouldn’t accept any edits to my genome without the consequences being very well understood (or in a last-ditch effort to save my life). I’d predict severe mental illness would happen alongside substantial intelligence gains.
Source: research career as a computational cognitive neuroscientist.
I put this as a post- ASI technology, but that’s also a product of my relatively short timelines.
Yes, I think many in the field would share this viewpoint and that’s part of why we haven’t seen someone already attempt this.
I disagree for reasons I’ve shared in my post on “Black Box Biology”, but it’s worth reiterating my reasons here:
You don’t need to understand the causal mechanism of genes. Evolution has no clue what effects a gene is going to have, yet it can still optimize reproductive fitness. The entire field of machine learning works on black box optimization.
Most genetic variants (especially those that commonly vary among humans, which are the ones we would be targeting) have linear effects on a single trait. We don’t actually need to worry about gene-gene interactions that much.
To the degree plieotropy does exist and is a concern, you can optimize your edit targeting criteria according to multiple traits. For example, you could try to edit to reduce (or at the very least keep constant) the risk of schizophrenia and other mental disorders.
(As stated in the post), a delivery vector that doesn’t induce an adaptive immune response can be administered in multiple rounds, with a relatively small number of edits made each time, further decreasing the risk of large side-effects.
As far as regulation goes, we’ve already approved one CRISPR-based gene therapy in the US. I see no reason to expect that you couldn’t conduct a clinical trial to treat a polygenic brain disease like Alzheimers or treatment resistant depression. That’s why in my roadmap I proposed clinical trails for treating a fatal brain disorder as a first step before we tackle intelligence.
Your point 2 is my big hangup. If you mean each genetic variant affects each trait roughly linearly, sure. If you mean each genetic variant affects only one trait, I think that’s completely wrong. Most studies only address the affect on a single trait, but given the re-use of proteins for different roles, I fully expect multiple effects of genes on average. It seems like I’ve seen studies and papers on this, but it’s never been my area, so I don’t remember anything clearly.
Evolution succeeds by tinkering over many generations. It creates as many downsides as upsides. Who’s going to volunteer to be tinkered upon?
Actually, as soon as I pose the question that way, I realize that the answer is “lots of people” (as long as there’s a reason to think you’ll get more upside than downside, which limited theory will provide.)
However, there’s no way the FDA is going to approve tinkering. You’d have to do this outside of US jurisdiction.
I am not saying plieotropy doesn’t exist. I’m saying it’s not as big of a deal as most people in the field assume it is.
Take disease risk for example. Here’s a chart showing the genetic correlations between various conditions:
With a few notable exceptions, there is not very much correlation between different diseases.
And to the extent that plieotropy does exist, it mostly works in your favor. That’s why most of the boxes are yellowish instead of bluish. Editing or selecting embryos to reduce the risk of one disease usually results in a tiny reduction of others.
Evolution cannot simultaneously consider data from millions of people when deciding which genetic variants to give someone. We can.
None of these proposals deal with novel genetic variants. Every target variant we would introduce is already present in tens of thousands of individuals and is known to not cause any monogenic disorder.
I’m not quite sure what you’re getting at here. Do you believe it’s impossible to make advantageous genetic tradeoffs? Or that there is no way to genetically alter organisms in a way that results in a net benefit?
The FDA routinely approves clinical trials to treat fatal diseases with no effective treatments. There are many lethal brain disorders that satisfy this requirement; Alzheimer’s, dementia, ALS, Parkinson’s and others.
Would the FDA approve a treatment to enhance intelligence? Probably not, unless US citizens were flying out of the country to get it. But if you can treat a polygenic brain disorder like Alzheimer’s with gene therapy, you can quite easily repurpose the platform to target intelligence by simply swapping the guide RNAs.
This matrix closes the case in my book
That matrix goes a long way in showing that there isn’t much correlation between diseases in the natural distribution. What is the reason to believe those correlations will remain low when you are making edits resulting in an extremely unlikely genome?
We’d edit the SNPs which have been found to causally influence the trait of interest in an additive manner. The genome would only become “extremely unlikely” if we made enough edits to push the predicted trait value to an extreme value—which you probably wouldn’t want to do for decreasing disease risk. E.g. if someone has +2 SD risk of developing Alzheimer’s, you might want to make enough edits to shift them to −2 SD, which isn’t particularly extreme.
You’re right that this is a risk with ambitious intelligence enhancement, where we’re actually interested in pushing somewhat outside the current human range (especially since we’d probably need to push the predicted trait value even further in order to get a particular effect size in adults) -- the simple additive model will break down at some point.
Also, due to linkage disequilibrium, there are things that could go wrong with creating “unnatural genomes” even within the current human range. E.g. if you have an SNP with alleles A and B, and there are mutations at nearby loci which are neutral conditional on having allele A and deleterious conditional on having allele B, those mutations will tend to accumulate in genomes which have allele A (due to linkage disequilibrium), while being purged from genomes with allele B. If allele B is better for the trait in question, we might choose it as an edit site in a person with allele A, which could be highly deleterious due to the linked mutations. (That said, I don’t think this situation of large-conditional-effect mutations is particularly likely a priori.)
Molecular biologists should be in charge of AI research and regulation, because AGI is not as big of a deal as AI researchers who work in the field assume it is.
The stakes could hardly be more different—polygenic trait selection doesn’t get everyone killed if we get it slightly wrong.
(I should clarify, I don’t see modification of polygenic traits just as a last ditch hail mary for solving AI alignment—even in a world where I knew AGI wasn’t going to happen for some reason, the benefits pretty clearly outweigh the risks. The case for moving quickly is reduced, though.)
Considering the enormity of my response, I figured I would post it in a place that is more visible to those interested. First I’d like to express my gratitude for you and GeneSmith’s goal and motivation; I agree that without some brain-machine interface solution, intelligence enhancement is certainly the way forward for us if we’d like to not only keep up with AI, but also break through the intellectual soft caps that have led to plateauing progress in many fields as they gradually become hyper-specialized. I don’t use this website very often, but fortunately this post was sent to me by a friend and I decided to engage with it since I had the exact same ideas and motivations detailed in your post when I matriculated into a PhD program for molecular genetics. I don’t want to de-anonymize myself, so I won’t mention the exact school but it is ranked within the top 50 graduate programs in the US and has had many Nobel Prize laureates as faculty. I was very optimistic, like you two appear to be. My optimism was short-lived and I eventually dropped out of the program because I quickly learned how infeasible this all was at the time (and still is), though, and figured I could make more of an impact by exerting my influence elsewhere. With that said, I’d like to amend what I said: I don’t believe that what you want to do as described in your post is impossible, but that it is infeasible.
The first impasse that I don’t see being mentioned much is gathering adequate sample data for building a useful predictor. As you stated yourself, “it is unlikely any of the groups that currently have the data to create strong intelligence predictors (or could easily obtain it) will do so.” The reality is even worse than this; not only will the industry’s magnates not build strong predictors despite their ability to, there are armies of fervent bureaucrats, lawyers, and researchers employed all across academia, health, and governments that will actively attempt to prevent this from transpiring, which is part of the reason why we don’t have it already. Even if this were not true and you could somehow orchestrate a collection of said data, it would take a very long time and a lot of money. To tease out a sufficient abundance of causal variants for intelligence as measured by an IQ test, my guess is you’d need around 500,000 samples at the very least. This guess is based on the findings of GWASes studying other complex traits, as well as the work of those at Genomic Prediction. This would first require that you find an overlap of test subjects five hundred thousand strong that will not only volunteer to have their entire genome sequenced (a SNP array could be used to cut costs if you’re willing to sacrifice the breadth of variants interrogated), but will also sit down for an hours-long professionally-administered IQ test, like the WAIS-IV (again, could use some abridged test to cut costs and increase participation rate at the expense of lower-quality data). Ignoring that this would take years because this population of 500,000 people would come from what I’d imagine is a very sparsely distributed subset of the broader population, this would be extremely expensive, as even the cheapest professional IQ tests cost at least $100 to administer, which already renders your project (or whatever entity funds these tests) at least $50,000,000 in the hole before you’ve even began designing and conducting experiments. You could elect to use proxy measures like educational attainment, SAT/ACT/GRE score, most advanced math class completed, etc., but my intuition is that they are influenced by too many things other than pure g to be useful for the desired purpose. It’s possible that I’m being too cynical about this obstacle and I would be delighted if someone could give me good reasons why I’m wrong.
The barriers involved in engineering the delivery and editing mechanisms are different beasts. HiddenPriors already did a satisfactory job at outlining these, though he could’ve been more elaborate. At the risk of being just as opaque, I will just give the bottom line, because a fully detailed explanation of why this is infeasible would require more text than I want to write. As far as delivery goes, the current state of these technologies will force you to use lipid nanoparticles because of the dangers of an inflammatory response being induced in the brain by an AAV, not to mention the risk of random cell death induction by AAVs, the causes of which are poorly understood. Your risk appetite for such things must be extremely low considering you do not want to sustain cell death in the brain if you want to keep subjects alive, never mind imparting intelligence enhancements. Finding an AAV serotype that does not carry these risks would be a breakthrough on its own, finding an AAV serotype or sufficiently abundant collection of AAV serotypes that are immune to neutralization following repeated exposures is another breakthrough, and finding an AAV that could encode all of the edits you want to make (obviating the need for multiple injections) is another breakthrough, and frankly is probably impossible. For the things that overcome this, you would still have to overcome the low transduction efficiency and massive costs of producing many different custom AAVs at scale, lest there is another breakthrough. As you mention, such challenges have a chance of being solved by the market eventually, though who knows when that will be if it ever happens at all. These would not be as big of issues as they are were you only wanting to make a few edits, but you want to make hundreds or thousands of edits, which necessitates using AAVs multiple times if you were to choose them which exponentially compounds the chance that these risks are realized. After having been in the field and witnessing how this type of research is performed, what goes on under the hood at research labs, how slow progress is, and the biochemistry of such vectors, my personal take on this matter is that attempting to solve all of these problems for AAVs is akin to trying to optimize a horse for the road when what you really need is a car, and that car is probably going to end up being lipid nanoparticles. They’re cheaper, safer, intrinsically much less immunogenic, and more capacious. You will need to use plasmid DNA (as opposed to mRNA, which is where lipid nanoparticles currently shine) if you want to keep them non-immunogenic and avoid the same immunogenicity risks of AAVs, which will significantly reduce your transduction efficiency lest you develop another breakthrough. Lipid nanoparticles, even though they’re generally much safer, still have the potential to be immunogenic or toxic following repeated doses or high enough concentrations, which is another hurdle because you will need to use them repeatedly considering the number of edits you’re wanting to make.
I have not even gotten to why base/prime editing as those methods exist will be problematic for the number of edits you’re wanting to make, but I will spare you because my rhetoric is getting repetitive; it basically boils down to what was already mentioned in your post, the replies, and the previous paragraph, that is that the more edits you make the much greater the chance that risks will be realized, in this case meaning things like pseudo-random off-target effects, bystander edits, and guiding RNA collisions with similar loci in the genome. I also disagree with both the OP and HiddenPriors regarding the likelihood that mosaicism will be a problem. A simple thought experiment may change your mind about mosaicism in the brain: consider what would happen in the case of editing multiple loci (whether purposeful or accidental) that happen to play a role in a neuron’s internal clock. If you have a bunch of neurons releasing substrates that govern one’s circadian rhythm in a totally discordant manner, I’d have to imagine the outcome is that the organism’s circadian rhythm will be just as discordant. This can be extrapolated to signaling pathways in general among neurons, where again one could imagine that if every 3rd or 4th or nth neuron is receiving, processing, or releasing ligands in a different way than either the upstream or downstream neurons, the result is some discordance that is more likely to be destructive than beneficial. Albeit an informed one, this is just a conjecture based on my knowledge of neurobiology and another case where I’d be delighted if someone could give me good reasons why I might be wrong.
For all of the reasons herein and more, it’s my personal prediction that the only ways humanity is going to get vastly smarter by artificial means is through brain machine interfaces or iterative embryo selection. There are many things in the OP that I could nitpick but that do not necessarily contribute to why I think this project is infeasible, and I don’t want to make this gargantuan reply any longer than it already is. I hope I wrote enough to give you a satisfactory answer for why I think this is infeasible; I would be glad to chat over email or discord if you would like to filter ideas through me after reading this.
I am more optimistic than you here. I think it is enough to get people who have already gotten their genomes sequenced through 23&Me or some other such consumer genomics service to either take an online IQ test or submit their SAT scores. You could also cross-check this with other data such people submit to validate their answer and determine whether it is plausible.
I think this could potentially be done for a few million dollars rather than 50. In fact companies like GenomeLink.io already have these kind of third party data analysis services today.
Also, we aren’t limited to western countries. If China or Taiwan or Japan or any other country creates a good IQ predictor, it can be used for editing purposes. Ancestry doesn’t matter much for editing purposes, only for embryo selection.
Would the quality of such tests be lower than those of professionally administered IQ tests?
Of course. But sample size cures many ills.
I briefly looked into this and found these papers:
Adeno-Associated virus induces apoptosis during coinfection with adenovirus
I asked GPT4 whether adenoviruses enter the brain:
I also found this paper indicating much more problematic direct effects observed in mouse studies:
AAV ablates neurogenesis in the adult murine hippocampus
Also:
So it sounds like there are potential solutions here and this isn’t necessarily a showstopper, especially if we can derisk using animal testing in cows or pigs.
This is an update for me. I didn’t previously realize that the mRNA for a base or prime editor could itself trigger the innate immune system. I wonder how serious of a concern this would actually be?
If it is serious, we could potentially deliver RNPs directly to the cells in question. I think this would be plausible to do with pretty much any delivery vector except AAVs.
I don’t really see how delivering a plasmid with the DNA for the editor will be any better than delivering mRNA. The DNA will be transcribed into the exact same mRNA you would have been delivering anyways, so if the mRNA for CRISPR triggers the innate immune system thanks to CpG motifs or something, putting it in a plasmid won’t help much.
Yeah, one other delivery vector I’ve looked into the last couple of days are extracellular vescicles. They seem to have basically zero problems with toxicity because the body already uses them to shuttle stuff around. And you can stick peptides on their surface similar to what we proposed with lipid nanoparticles.
The downside is they are harder to manufacture. You can make lipid nanoparticles by literally putting 4 ingredients plus mRNA inside a flask together and shaking it. ECVs require manufacturing via human cell colonies and purification.
Thanks for this example. I don’t think I would be particularly worried about this in the context of off-target edits or indels (provided the distribution is similar to that of naturally occuring mutations), but I can see it potentially being an issue if the intellligence modifying alleles themselves work via regulating something like the neuron’s internal clock.
If this turns out to be an issue, one potential solution would be to exclude edits to genes that are problematic when mosaic. But this would probably be pretty difficult to validate in an animal model so that might just kill the project.
I have experience attempting things like what you’re suggesting 23andMe do; I briefly ran a startup unrelated to genomics, and I also ran a genomics study at my alma mater. Both of these involved trying to get consumers or test subjects to engage with links, emails, online surveys, tests, etc., and let me be the first to tell you that this is hard for any survey longer than your average customer satisfaction survey. If 23andMe has ~14 million customers worldwide and they launch a campaign that aims to estimate the IQ scores of their extant customers using an abridged online IQ test (which would take at least ~15-20 minutes if it is at all useful), it is optimistic to think they will get even 140,000 customers to respond. This prediction has an empirical basis; 23andMe conducted a consumer experience survey in 2013 and invited the customers most likely to respond: those who were over the age of 30, had logged into their 23andMe.com account within the two‐year period prior to November 2013, were not part of any other 23andMe disease research study, and had opted to receive health results. This amounted to an anemic 20,000 customers out of its hundreds of thousands; considering 23andMe is cited to have had about ~500,000 customers in 2014, we can reasonably assume they had at least ~200,000 customers in 2013. To make our estimate of the invitation rate generous, we will say they had 200,000 customers in 2013, meaning 10% of their customers received an invitation to complete the survey. Even out of this 10%, slightly less than 10% of that 10% responded to a 98-question survey, so a generous estimate of how many of their customers they got to take this survey is 1%. And this was just a consumer experience survey, which does not have nearly as much emotional and cognitive friction dissuading participants as something like an IQ test. It is counterintuitive and demoralizing, but anyone who has experience with these kinds of things will tell you the same thing. If 23andMe instead asked customers to submit SAT/ACT/GRE scores, there are now many other problems to account for (other than a likely response rate of <=1% of total customer base): dishonest or otherwise unreliable reporting, selecting for things that are not intelligence like conscientiousness, openness, and socioeconomic status, the mean/standard deviation of scores being different for each year (so you’d have to calculate z-score differently based on the year participants took the tests), and the fact it is much easier to hit the ceiling on the SAT/ACT/GRE (2-3 S.D., 1 in 741 at the rarest) than it is to hit the ceiling of a reliable IQ test (4 S.D., which is about 1 in 30,000). Statistical models like those involved in GWASes follow one of many simple rules: crap in, crap out. If you want to find a lot of statistically significant SNPs for intelligence and you try using a shoddy proxy like standardized test score or an incomplete IQ test score as your phenotype, your GWAS is going to end up producing a bunch of shoddy SNPs for “intelligence”. Sample size (which is still an unsolved problem for the reasons aforementioned) has the potential to make up for obtaining a low amount of SNPs that have genome-wide significance, but it won’t get rid of entangled irrelevant SNPs if you’re measuring something other than straight up full-scale IQ. I really hate to be so pessimistic here, but it’s important to be realistic about these kinds of things especially if you’re relying on it to play a critical role in your project’s success.
Taiwan is one of the more tenable counter-examples I also thought of to what I said, but there are still problems to overcome. In the UK biobank for example, their method of assessing “fluid intelligence”/”verbal-numerical ability” was totally abysmal. They gave participants 2 minutes to answer 13 IQ-test-esque multiple-choice questions and their score was based on the number of questions they answered correctly in the 2 minutes. I hope I don’t need to explain why this is not an adequate measure of fluid intelligence and that any IQ predictor built on that data is probably totally useless. I don’t know how Taiwan assesses intelligence in their biobank if at all, but if they do it anything like how the UK biobank did it, that data will probably end up being similarly useless. Even after the fact, there is still the problem of inadequate sample size if it’s not half a million or more, and that it will take a long time for all of this to complete by my understanding. My ultimate prediction regarding this obstacle is that in order to build an IQ predictor in a short amount of time that has enough quality data to uncover a sufficient abundance of causal alleles for intelligence, there will need to be monetary incentives for the sought hundreds of thousands of participants, actual full-scale IQ tests administered, and full genome sequencing. Again, I would be delighted to be wrong about all of this and I encourage anyone to reply with good reasons for why I might be.
As mentioned in my reply, I would tend to agree if your goal was to only make a few edits and thus use an AAV only once or twice to accomplish this. This has been demonstrated to be relatively safe provided the right serotype is used, and there are even FDA-approved gene delivery therapies that use AAVs in the CNS. Even in these cases though, the risk of inducing an inflammatory response or killing cells is never zero even with correct dosing and single exposure, and for your purposes you would need to use at least hundreds of AAV injections to deliver hundreds of edits, and thousands of AAV injections to deliver thousands of edits. Again, barring some breakthrough in AAVs as delivery vectors, this number of uses in a single person’s CNS practically guarantees that you will end up inducing some significant/fatal inflammatory response or cytolysis. This is without even mentioning the problems of developing immunity to the viruses and low transduction efficiency, which are another couple of breakthroughs away from being solved.
You may find these two papers elucidating: one, two
This is interesting. This is the first time I’m hearing of these as they pertain to potential gene therapy applications. Here are some papers about them I found that you may find useful as you consider them as an option: one, two, three
To be candid with you, I was mostly just trying to play devil’s advocate regarding mosaicism. Like you mention, neurons accumulate random mutations over the lifespan anyways and it doesn’t seem to be detrimental necessarily, though one can’t disentangle the cognitive decline due to this small-scale mosaicism versus that due to aging in general. It’s also possible that having an order of magnitude increase in mosaicism (e.g., 1,000 random mutations across neurons to 10,000 random mutations across neurons) induces some phase transition in its latent perniciousness. Either way, if you solve either the transduction efficiency or immunological tolerance issues (if low transduction efficiency, just employ multiple rounds of the same edit repeatedly), mosaicism won’t be much of a problem if it was ever going to be one.
This is just measurement error and can be handled by normal psychometric approaches like SEM (eg. GSEM). You lose sample efficiency, but there’s no reason you can’t measure and correct for the measurement error. What the error does is render the estimates of each allele too small (closer to zero from either direction), but if you know how much error there is, you can just multiply back up to recover the real effect you would see if you had been able to use measurement with no error. In particular, for an editing approach, you don’t need to know the estimate at all—you only need to know that it is non-zero, because you are identifying the desired allele.
So, every measurement on every individual you get, whether it’s EA or SAT or GRE or parental degree or a 5-minute web quiz, helps you narrow down the set of 10,000 alleles that matters from the starting set of a few million. They just might not narrow it down much, so it becomes a more decision-theoretic question of how expensive is which data to collect and what maximizes your bang-for-buck. (Historically, the calculus has favored low-quality measurements which could be collected on a large number of people.)
The problem could potentially be solved by conducting GWASes that identify the SNPs of things known to correlate with the proxy measure other than intelligence and then subtracting those SNPs, but like you mention later in your reply, the question is what approach is faster and/or cheaper. Unless there is some magic I don’t know about with GSEM, I can’t see a convincing reason why it would have intelligence SNPs buoy to the top of lists ranked on the basis of effect size, especially with the sample size we would likely end up working with (<1 million). If you don’t know what SNPs contribute to intelligence versus something else, applying a flat factor to each allele’s effect size would just increase the scale of difference rather than help distill out intelligence SNPs. Considering the main limitation of this project is the number of edits they’re wanting to make, minimizing the number of allele flips while maximizing the effect on intelligence is one of the major goals here (although I’ve already stated why I think this project is infeasible). Another important thing to consider is that the p-value of SNPs’ effects would be attenuated as the number of independent traits affecting the phenotype increases; if you’re only able to get 500,000 data points for the GWAS that uses SAT as the phenotype, you will most likely have the majority of causal intelligence SNPs falling below the genome-wide significance threshold of p < 5 * 10E-8.
It’s also possible that optimizing peoples’ brains (or a group of embryos) for acing the SAT to the point where they have a 100% chance of achieving this brings us as close to a superintelligent human as we need until the next iteration of superintelligent human.
The tragedy of all of this is that it’s basically a money problem—if some billionaire could just unilaterally fund genome sequencing and IQ testing en masse and not get blocked by some government or other bureaucratic entity, all of this crap about building an accurate predictor would disappear and we’d only ever need to do this once.
More or less. If you have an impure measurement like ‘years of education’ which lumps in half intelligence and half other stuff (and you know this, even if you never have measurements of IQ and EDU and the other-stuff within individuals, because you can get precise genetic correlations from much smaller sample sizes where you compare PGSes & alternative methods like GCTA or cross-twin correlations), then you can correct the respective estimates of both intelligence and other-stuff, and you can pool with other GWASes on other traits/cohorts to estimate all of these simultaneously. This gets you estimates of each latent trait effect size per allele, and you just rank and select.
A statistical-significance threshold is irrelevant NHST mumbo-jumbo. What you care about is posterior probability of the causal variant’s effect being above the cost-safety threshold, whatever that may be, but which will have nothing at all to do with ‘genome-wide statistical significance’.
I’m aware of this, but if you’re just indiscriminately shoveling heaps of edits into someone’s genome based on a GWAS with too low a sample size to reveal causal SNPs for the desired trait, you’ll be editing a whole bunch of what are actually tags, a whole bunch of things that are related to independent traits other than intelligence, and a whole bunch of random irrelevant alleles that made it into your selection by random chance. This is a sure-fire way to make a therapy that has no chance of working, and if an indiscriminate shotgun approach like this is used in experiments, the combinatorics of the matter dictates that there are more possible sure-to-fail multiplex genome editing therapies than there are humans on the Earth, let alone those willing to be guinea pigs for an experiment like this. Having a statistical significance threshold imposes a bar to pass for SNPs that at least makes the therapy less of an ascertained suicide mission.
EDIT: misinterpreted what other party was saying.
What I said was “What you care about is posterior probability of the causal variant’s effect being above the cost-safety threshold”. If you are ‘indiscriminately shoveling’, then you apparently did it wrong.
Pretty much all SNPs are related to something or other. The question is what is the average effect. Given the known genetic correlations, if you pick the highest posterior probability ones for intelligence, then the average effect will be good.
(And in any case, one should be aiming for maximizing the gain across all traits as an index score.)
If they’re irrelevant, then there’s no problem.
No it’s not. If you’re using common SNPs which already exist, why would it ‘have no chance of working’? If some random SNP had some devastating effect on intelligence, then it would not be ranked high.
What if 23&me offered a $20 discount for uploading old SAT scores? I guess someone would set up a site that generates realistically distributed fake SAT scores that everyone would use. Is there a standardized format for results that would be easy to retrieve and upload but hard to fake? Eh, idk, maybe not. Could a company somehow arrange to buy the scores of consenting customers directly from the testing agency? Agree that this seems hard.
This seems unduly pessimistic to me. The whole interesting thing about g is that it’s easy to measure and correlates with tons of stuff. I’m not convinced there’s any magic about FSIQ compared to shoddier tests. There might be important stuff that FSIQ doesn’t measure very well that we’d ideally like to select/edit for, but using FSIQ is much better than nothing. Likewise, using a poor man’s IQ proxy seems much better than nothing.
I wouldn’t call it magic, but what makes FSIQ tests special is that they’re specifically crafted to estimate g. To your point, anything that involves intelligence (SAT, ACT, GRE, random trivia quizzes, tying your shoes) will positively correlate with g even if only weakly, but the correlations between g factor scores and full-scale IQ scores from the WAIS have been found to be >0.95, according to the same Wikipedia page you linked in a previous reply to me. Like both of us mentioned in previous replies, using imperfect proxy measures would necessitate multiplying your sample size because of diluted p-values and effect sizes, along with selecting for many things that are not intelligence. There are more details about this in my reply to gwern’s reply to me.
This may have missed your point, you seem more concerned about selecting for unwanted covariates than ‘missing things’, which is reasonable. I might remake the same argument by suspecting that FSIQ probably has some weird covariates too—but that seems weaker. E.g. if a proxy measure correlates with FSIQ at .7, then the ‘other stuff’ (insofar as it is heritable variation and not just noise) will also correlate with the proxy at .7, and so by selecting on this measure you’d be selecting quite strongly for the ‘other stuff’, which, yeah, isn’t great. FSIQ, insofar as it had any weird unwanted covariates, would probably much less correlated with them than .7
I might end up eating my words on the delivery problem. Something has just come out a few days ago that renewed a bit of my optimism, see here. According to the findings in this pre-print, it is possible to shield AAVs from the immune system using protein vaults that the immune system recognizes as self. It is not perfect though; although VAAV results in improved transduction efficiency even in the presence of neutralizing antibodies, it still only results in transduction of ~4% of cells if neutralizing antibodies are present. This means you’d need to cross your fingers and hope that 1) the patient doesn’t already have naturally extant neutralizing antibodies and 2) they don’t develop them over the course of the hundreds/thousands of VAAV you’re going to give them. In the paper, it is stated that AAV gets packaged in the vaults only to an extent rather than completely. So, more than likely, even if you’re injecting 99% VAAV and 1% naked AAV, if you do this 100 times you are almost sure to develop neutralizing antibodies to that 1% of naked AAV (unless they have a way to completely purify VAAV that removes all naked AAV). One way to combat the transduction problem post-innocuation though is using multiple injections of the same edit in order to approximate 100% transduction, though I’m pessimistic that this will work because there is probably a good reason that only 4% of cells were transducible; something might be different about them than the rest of cells, so you might receive diminishing transduction returns with each injection. They also still need to demonstrate that these work in vivo and that they can be routed to the CNS. Nonetheless, I’m excited to see how this shakes out.
Thanks for leaving such thorough and thoughtful feedback!
The SAT is heavily g-loaded: r = .82 according to Wikipedia, so ~2/3 of the variance is coming from g, ~1/3 from other stuff (minus whatever variance is testing noise). So naively, assuming no noise and that the genetic correlations mirror the phenotype correlations, if you did embryo selection on SAT, you’d be getting .82*h_pred/sqrt(2) SDs g and .57*h_pred/sqrt(2) SDs ‘other stuff’ for every SD of selection power you exert on your embryo pool (h_pred^2 is the variance in SAT explained by the predictor, we’re dividing by sqrt(2) because sibling genotypes have ~1/2 the variance as the wider population). Which is maybe not good; maybe you don’t want that much of the ‘other stuff’, e.g. if it includes personality traits.
It looks like the SAT isn’t correlated much with personality at all. The biggest correlation is with openness, which is unsurprising due to the correlation between openness and IQ—I figured conscientiousness might be a bit correlated, but it’s actually slightly anticorrelated, despite being correlated with GPA. So maybe it’s more that you’re measuring specific abilities as well as g (e.g. non-g components of math and verbal ability).
Another thing: if you have a test for which g explains the lion’s share of the heritable variance, but there are also other traits which contribute heritable variance, and the other traits are similarly polygenic as g (similar number of causal variants), then by picking the top-N expected effect size edits, you’ll probably mostly/entirely end up editing variants which affect g. (That said, if the other traits are significantly less polygenic than g then the opposite would happen.)
Getting old SAT scores could be much cheaper, I imagine (though doing this would still be very difficult). Also, as GeneSmith pointed out we aren’t necessarily limited to western countries. Assembling a large biobank including IQ scores or a good proxy might be much cheaper and more socially permissible elsewhere.
I do basically expect the delivery problem will gated by missing breakthroughs, since otherwise I’d expect the literature to be full of more impressive results than it actually is. (E.g. why has no one used angiopep coated LNPs to deliver editors to mouse brains, as far as I can find? I guess it doesn’t work very well? Has anyone actually tried though?)
Ditto for editors, though I’m somewhat more optimistic there for a handful of reasons:
sequence dependent off-targets can be predicted
so you can maybe avoid edits that risk catastrophic off-targets
unclear how big of a problem errors at noncoding target sites will be (though after reading some replies pointing out that regulatory binding sites are highly sensitive I’m a bit more pessimistic about this than I was)
even if they are a big problem, dCas9-based ABEs have extremely low indel rates and incorrect base conversions, though bystanders are still a concern
though if you restrict yourself to ABEs and are careful to avoid bystanders, your pool of variants to target has shrunk way down
I mean, your basic argument was “you’re trying to do 1000 edits, and the risks will mount with each edit you do”, which yeah, maybe I’m being too optimistic here (e.g. even if not a problem at most target sites, errors will predictably be a big deal at some target sites, and it might be hard to predict which sites with high accuracy).
It’s not clear to me how far out the necessary breakthroughs are “by default” and how much they could be accelerated if we actually tried, in the sense of how electric cars weren’t going anywhere until Musk came along and actually tried (though besides sounding crazy ambitious, maybe this analogy doesn’t really work if breakthroughs are just hard to accelerate with money, and AFAIK electric cars weren’t really held up by any big breakthroughs, just lack of scale). Getting delivery+editors down would have a ton of uses besides intelligence enhancement therapy; you could target any mono/oligo/poly-genic diseases you wanted. It doesn’t seem like the amount of effort currently being put in is concomitant with how much it would be worth, even putting ‘enhancement’ use cases aside.
My impression is neurons are really noisy, and so probably not very sensitive to small perturbations in timing / signalling characteristics. I guess things could be different if the differences are permanent rather than transient—though I also wouldn’t be surprised if there was a lot of ‘spatial’ noise/variation in neural characteristics, which the brain is able to cope with. Maybe this isn’t the sort of variation you mean. I completely agree that its more likely to be detrimental than beneficial, it’s a question of how badly detrimental.
Another thing to consider: do the causal variants additively influence an underlying lower dimensional ‘parameter space’ which then influences g (e.g. degree of expression of various proteins or characteristics downstream of that)? If this is the case, and you have a large number of causal variants per ‘parameter’, then if your cells get each edit with about the same frequency on average, then even if there’s a ton of mosaicism at the variant level there might not be much at the ‘parameter’ level. I suspect the way this would actually work out is that some cells will be easier to transfect than others (e.g. due to the geography of the extracellular space that the delivery vectors need to diffuse through), so you’ll have some cells getting more total edits than others: a mix of cells with better and worse polygenic scores, which might lead to the discordance problems you suggested if the differences are big enough.
BMI seems harder than in-vivo editing to me. Wouldn’t you need a massive number of connections (10M+?) to even begin having any hope of making people qualitatively smarter? Wouldn’t you need to find an algorithm that the brain could ‘learn to use’ so well that it essentially becomes integrated as another cortical area or can serve as an ‘expansion card’ for existing cortical areas? Would you just end up bottlenecked by the characteristics of the human neurons (e.g. low information capacity due to noise)?
The article that Wikipedia cites for that factoid, Frey & Detterman 2004, uses the National Longitudiunal Survey of Youth 1979 for its data that included the SAT and ASVAB (this is what they used to estimate IQ, so first need to find correlation between ASVAB and actual FSIQ) scores for the samples. This introduces the huge caveat that the SAT has changed drastically since this study was conducted and is likely no longer nearly as strongly correlated with g ever since 1994. This is when they began recentering scores and changing the scoring methodology, making year-to-year comparisons of scores no longer apples to apples. The real killer was their revision of the math and verbal sections to mostly include questions that “approximate more closely the skills used in college and high school work”, get rid of “contrived word problems” (e.g., the types of verbal ability questions you’d see on an IQ test), and include “real-world” problems that may be more relevant to students. Since it became more focused on assessing knowledge rather than aptitude, this rehauling of the scoring and question format made it much more closely reflect a typical academic benchmark exam rather than an assessment of general cognitive ability. This decreased its predictive power for general intelligence and increased its predictive power for high school GPA, as well as other things that correlate with high school GPA like academic effort, openness, and SES. It’s for these reasons that Mensa and other psychometrics societies stopped using SAT as an acceptable proxy for IQ unless you took it prior to 1994. I’ve taken both the SAT and ACT and I cannot imagine the ACT is much better (2004 study showed r=0.73). My guess is that the GRE would be much more correlated with general intelligence than either of the other two tests (still imperfectly so, wouldn’t put it >0.8), but then the problem is that a much smaller fraction of the population has taken the GRE and there is a large selection bias as to who takes it. Same with something like the LSAT. I still think the only way you will get away with cheaply assessing general intelligence is via an abridged IQ test such as that offered by openpsychometrics.org if it was properly normed and made to be a little longer.
I agree, but then you’re limiting yourself to whatever number of polymorphisms are left over after what is presumably a pseudo-arbitrary threshold, and you’d need a much larger sample size because the effect sizes and p-values of SNPs would be diluted because you’d now have many more polymorphisms contributing to the phenotype. Like you suggest, it is also a large inferential leap to assume this would exclusively result in variants that affect g. Refer to my reply to gwern for more about this.
Refer to the first paragraph of this reply and my reply to GeneSmith.
I agree, I think the delivery problem is a much taller mountain to climb than the editor problem. One of the reasons for this is the fact that editing is generally a tractable organic chemistry problem and delivery is almost exclusively an intractable systems biology problem. Considering the progress that precision genome editing tools have made in the past 10 years, I think it is reasonable to rely on other labs to discover ways to shave down the noxious effects of editing alone to near negligibility.
As you alluded to, the difference is that one thing was basically solved already. Making leaps forward in biology requires an insane amount of tedium and luck. Genius is certainly important too, but like with the editing versus delivery tractability problem, engineering things like batteries involves more tractable sub-problems than getting things to work in noisy, black box, highly variable wetware like humans.
Frankly, I know much less about this topic than the other stuff I’ve been talking about so my opinions are less strong for BMIs, but what has made me optimistic about such things is the existence of brain implants that have cured peoples’ depression, work showing that transcranial magnetic stimulation has the potential to enhance certain cognitive domains, and existing BMIs that cure paralysis at the level of the motor cortex. Like other things I mentioned, this also seems like somewhat of a more tractable problem, considering computational neuroscience is a very math intensive field of study and AI has vast potential to assist us in figuring it out. If the problem eventually comes down to needing more and more connections, I cannot imagine it will remain a problem for long, since it sounds relatively easier to figure out how to insert more fine connections into the brain than the stuff we’ve been discussing.
I should mention, when I wrote this I was assuming a simple model where the causal variants for g and the ‘other stuff’ are disjoint, which is probably unrealistic—there’d be some pleiotropy.
If the mechanism of most of these genes is that their variants push something analogous to a hyperparameter in one direction or the other, and the number of parameters is much smaller than the number of genes, then this strategy will greatly underperform the simulated prediction. This is because the cumulative effect of flipping all these genes will be to move hyperparameters towards optimal but then drastically overshoot the optimum.
If you were to flip enough variants to push someone far outside the human range then that’s almost certainly correct. But the linear model holds remarkably well within the current human range, and likely to some degree outside of it.
But I am not too concerned about this because we can do multiples rounds of fewer edits and validate between rounds.
I’d be worried about changes to my personality or values from editing so many brain relevant genes.
Fair. I doubt there would be THAT much value drift due to personality changes just because there’s not that much plieotropy in the genome. But it also wouldn’t be literally zero.
And you could always opt for a lesser effect size by just targeting fewer genes and testing yourself after some time period to assess values drift before deciding if you want to go forward with a larger intervention.
That is one of the nice things about this proposal; if it works well you could will be able to customize the effect size.
Thanks, this is very interesting.
One thing I don’t understand: you write that a major problem with viruses is:
Is this a problem for people who expect to only want one genetic modification during their lifetime?
So there are two separate concerns:
One is a concern for people who are getting a single dose monogenic gene therapy who already have antibodies to an AAV delivery vector due to a natural infection. In these cases, doctors can sometimes switch the therapy to use an AAV with a different serotype that can’t be attacked by the patient’s existing antibodies. If that’s not available, they’ll sometimes give patients immunosupressants.
The problem is more relevant in the context of multiplex editing because you may not be able to make all the edits you’d like to in one round of therapy. You can only inject so many AAVs or lipid nanoparticles or what have you at a time. Cells only have a limited capacity to process and break down editor proteins, and other waste products of the editing process. So you may need to do multiple editing rounds to achieve the desired effect.
It will be a lot easier to do this if the delivery vector itself doesn’t trigger the immune system. If it does, antibodies formed during the first round of edits will attack and destroy the the delivery vector. Maybe you can just give someone immunosupressants during each round of treatment? Or maybe you can just use a different AAV? There are potential solutions but I don’t yet know which are likely to work best.
Repeat administration is a problem for traditional gene therapy too, since the introduced gene will often be eliminated rather than integrated into the host genome.
Consolidating my previous comments:
I discussed this project with GeneSmith and I think it is promising, though very challenging to implement in practice. The hardest part will be safely and efficiently delivering the editing agent to a large fraction of the cells in the brain.
Some other points:
This makes it sound like CAR-T is gene editing, but it isn’t. Instead of editing a gene, it introduces a new one (a chimeric T-cell receptor). Although some companies are working on gene editing to enhance CAR-Ts.
The paper reporting this was here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7229841/
Thanks! I’ve added a link to the paper by Cory Smith and others to the relevant section and updated the section about CAR T-cell therapy to read “adds a single gene” rather than “modifies a single gene”
If anyone is interested, I’m working on conducting small-scale, informal random and controlled trials for various nootropics that improve neuroplasticity and IQ, from dihexa to anthocyanin. The medicine I want to test has documented effects, but I want to gather more data on what mental subskills they improve performance on the most and how strong the effect is.
In order to aid recruitment, every participant will get to take the intervention, just in a staggered order.
I have several people who’ve agreed to participate. However, I’ve paused the initiative to focus on other projects. Let me know if you want to take over as lead researcher! The mesh will thank you.
Hello, I am willing to take part in the study. Could you write to me privately?
If you get massively multiplex editing working in a dish, there are a bunch of studies these days exposing neurons in a dish to stimuli and training them to play some game or do some task.
Imagine if over 100 dishes of edited neurons you saw a slight (statistically significant) increase in performance.
Also, don’t sleep on the astrocytes.
That’s an interesting idea for validation, thanks.
I think this is a very important question to ask.
Sometimes the reasons are encouraging:
An inadequacy analysis reveals that no one is properly incentivized to.
The funding isn’t there.
It’s too schelp-y and unsexy.
It’s too science fiction-y.
No one thought of it.
Other times the reasons are discouraging:
There are good technical reasons.
There are annoying roadblocks that await. Maybe legal things. Things that are actually really difficult to bypass.
These definitely aren’t exhaustive lists. It’s just what came to me after a few minutes of thinking.
I see this as a sort of “stand on the shoulders of giants” type of situation. By consulting with domain experts and figuring out why people currently aren’t doing this, you’re standing on their shoulders. Once you’re up there and have a better view of the path forward:
You might want to continue down the path you’re currently headed.
You might see some roadblocks and realize you need to take a different path forward.
You might realize that it’s not worth moving forward at all.
I’m glad to see that you’ve put some solid thought into this question. However, I was concerned to see this:
That feels to me like you’re standing on their ankles[1] rather than their shoulders. The comment from HiddenPrior also gives me that sense.
To me, it seems worth spending a ton of time talking to people to really ensure that you are in fact on their shoulders.
I very well may be wrong about where it is exactly that you’re standing though. I only gave this post and the comment section a somewhat brief skim. It also seems plausible that you undersold just how much effort you’ve spent trying to stand on the shoulders of others.
The metaphor certainly breaks down, but I think the point still is clear.
I think at this point we’ve probably spoken with about 10 people I consider to have some reasonable level of expertise in the field. And there have been a number of very high quality comments from knowledgeable people in the comments.
We will continue to talk with more and perhaps we will learn something that will definitely not work. I think if that is the case, the polygenic editing path is still worth pursuing because it could potentially be repurposed for many other applications.
Promoted to curated: I am quite excited about this. If indeed adult gene editing is possible, this does open up a huge range of possibilities, and seems like a very important development to track. This post is also quite well written, and explains a bunch of important concepts around gene editing and intelligence and the relationship between genes and high-level traits of an organism pretty well.
Could someone open a manifold market on the relevant questions here so I could get a better sense of the probabilities involved? Unfortunately, I don’t know the relevant questions or the have the requisite mana.
Personal note- the first time I came into contact with adult gene editing was the youtuber Thought Emporium curing his lactose intolerance, and I was always massively impressed with that and very disappointed the treatment didn’t reach market.
This may be far future, but what do you think of Fanzors over CRISPRs?
Also Minicircles?
The smaller size of Fanzors compared to Cas9 is appealing and the potential for lower immunogenicity could end up being very important for multiplex editing (if inflammation in off-target tissues is a big issue, or if an immune response in the brain turns out to be a risk).
The most important things are probably editing efficiency and the ratio of intended to unintended edits. Hard to know how that will shake out until we have Fanzor equivalents of base and prime editors.
I hadn’t even heard of Fanzors before you mentioned them. Very interesting.
So it’s basically an endonuclease native to eukaryotes! After a brief search I haven’t been able to find any papers in which it has been used as an editor.
The best case scenario here would be that the cieling for editing efficiency and specificity for Fanzor-based editors would be higher than for CRISPR-based alternatives. I am unsure at the moment whether that’s true, and we likely won’t know for a little while.
Minicircles are cool, but they just seem like… small plasmids? Granted I am not an expert on this topic and they definitely have advantages over SOME types of plasmids, such as those that contain CpG sequences, which can trigger TLR9 activation and get the immune system riled up.
I mean they’re great in the sense that a plasmid that doesn’t contain bacterial genes and can fit in a delivery vector like an AAV are great. I just don’t really see them as a separate category of thing we haven’t had before.
https://a16z.com/announcement/investing-in-tome-biosciences/
Molecular Biologist here. Promoters (and any non-coding regulatory sequence for that matter) are extremely sensitive to point mutations. Since their sequence determines how well the RNA-polymerase binds to them, any change in the sequence of bind motifs or even in the distance between these motifs has a major (generally negative) impact in transcription initiation efficiency. https://www.nature.com/articles/s41580-018-0028-8
In fact, there is a whole field of research based on randomizing certain parts of a promoter to create a library with different properties/strength.
Well known library for bacterial promoters: https://parts.igem.org/Promoters/Catalog/Anderson
more info on promoter libraries: https://sci-hub.se/https://pubs.acs.org/doi/full/10.1021/acssynbio.8b00115
A really important question here is whether the causal SNPs that affect polygenic traits tend to be located in these highly sensitive sequences. One hypothesis would be that regulatory sequences which are generally highly sensitive to mutations permit the occasional variant with a small effect, and these variants are a predominant influence on polygenic traits. This would be bad news for us, since even the best available editors have non-negligible indel rates at target sites.
Another question: there tend to be many enhancers per gene. Is losing one enhancer generally catastrophic for the expression of that gene?
Thanks for the comment. This is actually quite helpful, as the effects of off-target edits or indels to promoter and enhancer regions is one of the primary uncertainties we have regarding feasibility of the proposal.
My prior for thinking that a few off-targets targets or indels wouldn’t necessarily be catastrophic was a paper I read that looked at the total accumulation of random mutations to neurons over the lifespan. I believe by age 40 the average person has about 1500.
Regulatory regions make up about 2% of the genome, so the average neuron has about 30 mutations in regulatory regions by the age of 40. So if we can keep our de novo mutations from increasing that number very much it will probably be ok.
Now it’s possible that the types of errors introduced by random mutations are of a different kind than those introduced by indels and off-targets from base and prime editors. A quick google search reveals that most de novos seem to be single base pair changes rather than insertion or deletion errors. So perhaps it WILL be an issue, at least for some editor variants.
I think the ideal approach to answer this question would be to use (or make) a computational model to predict the distribution of off-target edits and indels from editor variants, another to predict binding affinity as a function of sequence, and see how strongly such errors affect binding affinity. We could then compare those results to the affects of binding affinity from de novo mutations to see whether they were comparable in magnitude.
Perhaps others have already made such models. A quick search didn’t turn up anything, but I will continue looking.
The other option is to just test it empirically in cell cultures and then animal models.
If you have any other advice about how to approach this problem, I’d appreciate it.
I agree that in the grand scheme of things it would probably not make much of a difference. Also your 2% estimation is generous, if you consider that in any differentiated human cell most of the genes are inactivated. Mutations on those genes would thus be harmless
There’s a great youtuber called the thought emporium who did genetic engineering on himself. I highly recommend checking them out :
https://www.youtube.com/watch?v=J3FcbFqSoQY
And the 2year follow up: https://www.youtube.com/watch?v=aoczYXJeMY4
The tldr is he created a virus then ate it to make his digestive system have more of the gene that makes lactase as he was very intolerant. 2 years later the effects are starting to wear off as cells get replaced but it seems to have had a very high ROI
None of the stuff that you suggested has worked for any animal. I’m not saying it’s impossible, but it is far harder to achiever compared to the stuff that HAS been demonstrated on mice.
I am PhD in Bio, have an extensive experience with stem cells and gene editing. The idea of human/animal cognitive enhancement is great, but the delivery of gene therapy to adult brains is doomed: first, it’s technically challenging if not impossible, second, if we want to achieve a true revolution in cognition, we need to target brain development not already developed brain!
Imagine a monkey thinking of enhancing its abilities by injecting virus in its brain—will it ever reach a human level cognition? Sounds laughable. Who cares about +5 points to IQ, I want to see a 10xEinstein ;)
For >40 years, way before the discovery of CRISPRs and base editors, we’ve been successfully genetically engineering mice, but not other species. Why only mice? Because we can culture mouse embryonic stem cells that can give rise to complete animals. We did not understand why mouse cells were so developmentally potent, and why this didn’t work for other species. Now we do (I’m the last author):
Highly cooperative chimeric super-SOX induces naive pluripotency across species—ScienceDirect
When you engineer stem cells rather than adult animals, all of those concerns you listed are gone: low efficiency, off-target mutations, delivery, etc. Pluripotent stem cells are immortal and clonogenic, which means that even if you get 1 in 1000 cells with correct edits and no off-target mutations, you can expand it indefinitely, verify by sequencing, introduce more edits, and create as many animals as you want. The pluripotent stem cells can either be derived from the embryos or induced artificially from skin or blood cells. The engineered pluripotent stem cells can either be used directly to create embryos or can be used to derive sperm and eggs; both ways work well for mice.
Of course, attempting to engineer human babies right away is illegal and irresponsible. One would need to start with animals. I propose starting with rats, which are a great model of cognitive studies. Then move to dogs, for which training protocols are well-established, and therefore the cognition and trainability can be easily evaluated. Besides, enhanced dogs will have a commercial value (military, service dogs, etc.), generating cash which can be used for R&D. The IP generated for dogs could one day be applied to other species, including primates and eventually ourselves. While I agree that going for natural variations for human is the safest choice, I believe that to achieve a real cognitive revolution well beyond current human abilities, we might need to target general mechanisms of cognition, for example by engineering “super” versions of human brain master regulators, such as FOXP2, ARHGAP11B, etc.:
Human-specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset | Science
A humanized version of Foxp2 affects ultrasonic vocalization in adult female and male mice—von Merten − 2021 - Genes, Brain and Behavior—Wiley Online Library
Has anyone done 2500 edits in the brain cells of an animal? No. The graphs are meant to illustrate the potential of editing to affect IQ given a certain set of assumptions. I think there are still significant barriers that must be overcome. But like… the trend here is pretty obvious. Look at how much editors have improved in just the last 5 years. Look at how much better our predictors have gotten. It’s fairly clear where we are headed.
Also, to say that none of this stuff has been done in animals seems a bit misleading. Here’s a paper where the authors were able to make a desired edit in 60% of mouse brain cells. Granted, they were using AAVs, but for some oligogenic conditions that may be sufficient; you can pack a single AAV with a plasmid holding DNA sufficient to make sgRNA for 31 loci using base editors. There are several conditions for which 30 edits would be sufficient to result in a >50% reduction in disease risk even after taking into account uncertanties about which allele is causal.
Granted, if we can’t improve editing efficiency in neurons to above 5% then the effect will be significantly reduced. I guess I am fairly optimistic on this front: if an allele is having an effect in brains, it seems reasonable to assume that some portion of the time it will not be methylated or wrapped around a histone, and thus be amenable to editing.
Regarding lipid nanoparticles as a delivery vehicle for editors: Verve-101 is a clinical trial underway right now evaluating safety and efficacy of lipid nanoparticles with a base editor to target PCSK9 mutations causing familial hypercholesterolemia.
There are other links in the post such as one showing transcytosis of BBB endothelial cells using angiopep conjugated LNPs. And here’s a study showing about 50% transfection efficiency of LNPs to brain cells following intracranial injection in mice.
Technically challenging? Yes.
Impossible?
Obviously not. You can get payloads into the brain. You can make edits in cells. And though there are issues with editing efficiency and delivery, both continue to improve every year. Eventually we will be able to do this.
If your contention is that it is easier to get a large effect by editing embryos vs the adult brain, I would of course agree! But consider all the conditions that are modulated by the timing and level of protein expression. It would be quite surprising to me if intelligence were not to modulated in a similar manner.
Furthermore, given what is happening in AI right now, we probably don’t have 25 years left for the technology for embryo editing to mature and for the children born with its benefits to grow up.
I have doubts we can enhance chimpanzee intelligence. We don’t have enough chimpanzees or enough intelligence phenotypes to create GWAS for chimp intelligence (or any other mental trait for that matter).
We could try porting human predictors but well… we already see substantial dropoff in variance explained when predictors are ported from one genetic ancestry group to another. Imagine how large the dropoff would be between species.
Granted, a lot of the dropoff seems to be due to differences in allele frequencies and LD structure. So maybe there’s some chance that a decent percentage of the variants would cause similar effects across species. But my current guess is few of the variants will have effects in both species.
Also, if I expected +5 IQ points to be the ceiling of in-vivo editing I wouldn’t care about this either. I do not expect that to be the ceiling, which is reflected in some of the later graphs in the post.
I’ve spent the better part of the afternoon reading and trying to understand this paper.
First, it’s worth saying just how impressive this work is. The improvement of success rates over existing embryogenesis techniques like SCNT. I have a few questions I wasn’t able to find answers to in the paper:
Do the rates of full-term and adult survival rates in iPSC mice match that which could be achieved by normal IVF, or do they indicate that there is still some suboptimality in culturing of tetraploid aggregated iPSC embryos? I’m not familiar with the normal rates of survival for mice so I wasn’t able to tell from the graph whether there is still room for improvement.
How epigenetically different are embryos produced with Sox2-17 compared to those produced through the normal IVF process?
If this process or an improved one in the future were capable of inducing embryo-viable iPSC’s, would you be able to tell this was the case in humans with the current data available? If not, what data are you missing? I’m particularly wondering about whether you feel that there is sufficient data available regarding the epigenetic state of normal embryonic cells at the blastocyst stage.
You are of course correct about everything here. And if we had unlimited time I think the germline editing approach would be better. But AGI appears to be getting quite near. If we haven’t alignment by the point that AI can recursively self-improve, then I think this technology becomes pretty much irrelevant. Meat-based brains, even genetically enhanced ones, are going to be irrelevant in a post-AGI world.
How exactly do you propose to do this given we don’t have cognitive ability GWASes for rats, don’t have a feasible technique for getting them without hundreds of thousands of phenotypes, and given the poor track record of candidate gene studies in establishing causal variants?
Do the rates of full-term and adult survival rates in iPSC mice match that which could be achieved by normal IVF, or do they indicate that there is still some suboptimality in culturing of tetraploid aggregated iPSC embryos? I’m not familiar with the normal rates of survival for mice so I wasn’t able to tell from the graph whether there is still room for improvement.
Using tetraploid complementation, it is possible to achieve up to 70% of full-term development, which is similar rate of mouse natural conception. And this was before we understood how it works. I believe that soon we will be able to outperform nature and achieve close to 100% full term development and survival (I’ve seen 90% efficiency in some experiments). For human, only 30% of naturally conceived embryos are born, and only 10% of IVF, so superseding nature for human will be even easier than for mice.
How epigenetically different are embryos produced with Sox2-17 compared to those produced through the normal IVF process?
In figure 4 we demonstrate that the mice are healthy, and can breed giving rise to healthy progeny, which is the highest bar for the quality of the cells. Again, our current IVF practice has only 10% success rate—the bar is pretty low. Also, the biggest advance of the paper is not creation of Sox2-17, but understanding the mechanism of naive pluripotency in mammals, which gives the unprecedented access to mammalian germline. Before, it was only accessible for mice and rats.
If this process or an improved one in the future were capable of inducing embryo-viable iPSC’s, would you be able to tell this was the case in humans with the current data available? If not, what data are you missing? I’m particularly wondering about whether you feel that there is sufficient data available regarding the epigenetic state of normal embryonic cells at the blastocyst stage.
This is just the first paper on the true nature of naive cells. Mouse is always first. The paper is unusual in the way that it contains 4 more species, including human. The next step would be to achieve tetraploid complementation for non-rodents, such as pigs, cows, sheep, dogs, monkeys, etc. If we could generate various animals and they are heathy and give normal progeny, then only we could think of humans. For humans, the first edits will address horrendous genetic diseases, rather than enhancements.
FYI, your iPSCs would give rise to your clones rather than children, which might only be okay for individuals with high value for society (eg. Einstain-like intelligence). I think it makes more sense to derive ESCs from IVF embryos, edit them in the dish, do QC, then use to create the embryos again—those will obviously be your children. Another option is to use iPSCs for in vitro gametogenesis (IVG), so basically your edited iPSCs are used to derive sperm/eggs. This rout will take longer to perfect, because so far very few mice have been born from IVG.
Do you know a study that has demonstrated enhancement of intelligence by editing adults? It would be a cool study, definitely worth to pursue, but there’s a big change it won’t work at all. I would bet on cell therapy for adults rather than gene therapy.
On the other hand, multiple studies have already shown enhanced intelligence for mice and monkeys by engineering the germline.
AGI will hopefully not kill all the humans. With such pessimism we can just give up and watch tv. If there are any humans in future it makes sense to enhance their intelligence and other talents. I did not suggest enhancing monkeys, I was just trying to say that if we want to achieve a chimp-to-human level transformation for human, we need to target the development.
I am a relatively recently reformed geneticist/molecular biologist and previously used CRISPR/Cas9 at the bench in an experimental context. I no longer work in the lab and admit am not well-read on the latest literature.
I think this approach is interesting, and theoretically executable, but practically infeasible at the current maturity level of the relevant technologies. I’m not sure such a mission would be a good use of expertise and money at this stage. I share the views of a lot of the top level commenters here about the limited feasibility of the approach on both scientific and societal grounds.
I will not repeat the concerns made by others but have two related comments which, while they have been touched on by yourself and others already, hopefully add to the discussion:
1. I am skeptical of claims that editing techniques have zero/minimal/negligible off-target effects generally, and think you need to have an exceptionally, exceptionally strong evidence base to support this before getting near humans, considering the volume of edits you need to make.
You acknowledge this but I feel you downplay the risk of cancer—an accidental point mutation in a tumour suppressor gene or regulatory region in a single founder cell could cause a tumour. Historically gene therapies (e.g for x-linked SCID) have had issues with this in the past. While the mechanics of this are different, there are obviously similarities which would make me (and any regulatory authority) extremely cautious.
2. You propose that variants in non-coding regions would be preferable to target, since off-target mutations would have less effect in non-coding regions.
Firstly, if you’re this worried about off-targets/incorrect edits then this is quite a major concern for the feasibility of the approach (see #1 above). If you’re not actually worried about off-targets/incorrect edits then you should surely be confident to target coding regions?
Secondly, I don’t follow the logic that choosing non-coding targets would be safer as this would lead to only non-coding off-target mutations. As far as I am aware off-target mutations can take place anywhere in the genome, and it is not the case that having a target in a non-coding region would mean that off-targets were also in non-coding regions. [Unless you are using the term “off-target” to refer to any incorrect edit of the target site, and wider unwanted edits—in my community this term referred specifically to ectopic edits elsewhere in the genome away from the target site.]
I wondered if this assertion was based on there being evidence that editing of the type you propose to use has a greater risk of off-target mutations the closer you are to the target site? But even if that is the case, non-coding and coding regions are adjacent to each other in the genome so a nearby mutation could just as well affect a coding region.
For each target the likely off-targets can be predicted, allowing one to avoid particularly risky edits. There may still be issues with sequence-independent off-targets, though I believe these are a much larger problem with base editors than with prime editors (which have lower off-target rates in general). Agree that this might still end up being an issue.
This is exactly it—the term “off-target” was used imprecisely in the post to keep things simple. The thing we’re most worried about here is misedits (mostly indels) at noncoding target sites. We know a target site does something (if the variant there is in fact causal), so we might worry that an indel will cause a big issue (e.g. disabling a promoter binding site). Then again, the causal variant we’re targeting has a very small effect, so maybe the sequence isn’t very sensitive and an indel won’t be a big deal? But it also seems perfectly possible that the sequence could be sensitive to most mutations while permitting a specific variant with a small effect. The effect of an indel will at least probably be less bad than in a coding sequence, where it has a high chance of causing a frameshift mutation and knocking out the coded-for protein.
The important figure of merit for editors with regards to this issue is the ratio of correct edits to misedits at the target site. In the case of prime editors, IIUC, all misedits at the target site are reported as “indels” in the literature (base editors have other possible outcomes such as bystander edits or conversion to the wrong base). Some optimized prime editors have edit:indel ratios of >100:1 (best I’ve seen so far is 500:1, though IIUC this was just at two target sites, and the rates seem to vary a lot by target site). Is this good enough? I don’t know, though I suspect not for the purposes of making a thousand edits. It depends on how large the negative effects of indels are at noncoding target sites: is there a significant risk the neuron gets borked as a result? It might be possible to predict this on a site-by-site basis with a better understanding of the functional genomics of the sequences housing the causal variants which affect polygenic traits (which would also be useful for finding the causal variants in the first place without needing as much data).
Good post. This looks possible, if not feasible.
“crazy, unpredictable, and dangerous” are all “potentially surmountable issues”. It’s just that we need more research into them before they stop being crazy, unpredictable, and dangerous. (except quantum I guess)
I think that most are focusing on single-gene treatments because that’s the first step. If you can make a human-safe, demonstrably effective gene-editing vector for the brain, then jumping to multiplex is a much smaller step (effective as in does the edits properly, not necessarily curing a disease). If this were a research project I’d focus on researching multiplex editing and letting the market sort out vector and delivery.
I am more concerned about the off-target effects; neurons still mostly function with a thousand random mutations, but you are planning to specifically target regions that have a supposed effect. I would assume that most effects in noncoding regions are regulator binding sites (alternately: ncRNA?), which are quite sensitive to small sequence changes. My assumption would be a higher likelihood of catastrophic mutations (than you assume).
Promoters have a few of important binding motifs whose spacing is extremely precise, but most of the binding motifs are a lot more flexible in how far away they are from each other.
Also, given that your target is in nonreplicating cells, buildup of unwanted protein might be an issue if you’re doing multiple rounds of treatment.
The accuracy of your variant data could/should be improved as well; most GWAS-based heritability data assumes random mating which humans probably don’t do. But if you’re planning on redoing/rechecking all the variants that’d be more accurate.
Additionally, I’m guessing a number of edits will have no effect as their effect is during development. If only we had some idea how these variants worked so we can screen them out ahead of time. I’m not sure what percent of variants would only have an effect during development, so you’ll need to do a lot more edits than strictly necessary and/or a harder time detecting any effects of the edits. Luckily, genes that are always off are more likely to be silenced, so they might be harder to edit.
Though I would avoid editing unsilenced genes anyways, because they’re generally off and not being expressed (and therefore less likely to have a current effect) and the act of editing usually unsilences the genes for a bit, which is an additional level of disruption you probably don’t want to deal with.
I don’t know how the Biobank measures “intelligence” but make sure it corresponds with what you’re trying to maximize [insert rehash of IQ test accuracy].
Finally, this all assumes that intelligence is a thing and can be measured. Intelligence is probably one big phase space, and measurements capture a subset of that, confounded by other factors. But that’s getting philosophical, and as long as it doesn’t end up as eugenics (Gattaca or Hitler) it’s probably fine.
Honestly just multiplex editing by itself would be useful and impressive, you don’t have to focus on intelligence. Perhaps something like muscle strength or cardiovascular health would be an easier sell.
Makes sense.
The thing we’re most worried about here is indels at the target sites. The hope is that adding or subtracting a few bases won’t be catastrophic since the effect of the variants at the target sites are tiny (and we don’t have frameshifts to worry about). Of course, the sites could still be sensitive to small changes while permitting specific variants.
I wonder whether disabling a regulatory binding site would tend to be catastrophic for the cell? E.g. what would be the effect of losing one enhancer (of which there are many per gene on average)? I’d guess some are much more important than others?
This is definitely a crux for whether mass brain editing is doable without a major breakthrough: if indels at target sites are a big deal, then we’d need to wait for editors with negligible indel rates (maybe ≤10−5 per successful edit, while the current best editors are more like 10−3 to 10−2).
If the degradation of editor proteins turns out to be really slow in neurons, we could do a lower dose and let them ‘hang around’ for longer. Final editing efficiency is related to the product of editor concentration and time of exposure. I think this could actually be a good thing because it would put less demand on delivery efficiency.
Studying the transciptome of brain tissue is a thing. That could be a way to find the genes which are significantly expressed in adults, and then we’d want to identify variants which affect expression of those genes (spatial proximity would be the rough and easy way).
Significant expression in adults is no guarantee of effect, but seems like a good place to start.
g sure seems to be a thing and is easy to measure. That’s not to say there aren’t multiple facets of intelligence/ability—people can be “skewed out” in different ways that are at least partially heritable, and maintaining cognitive diversity in the population is super important.
One might worry that psychometric g is the principal component of the easy to measure components of intelligence, and that there are also important hard to measure components (or important things that aren’t exactly intelligence components / abilities, e.g. wisdom). Ideally we’d like to select for these too, but we should probably be fine as long as we aren’t accidentally selecting against them?
A huge and fascinating topic. But… I find myself thinking: suppose I wanted to change the color of my eyes. I could figure out how to gene-hack my iris—or I could get colored contact lenses.
If the objective is to make people smarter, to what extent can this be accomplished by being specific about the cognitive skills that are to be enhanced, and then identifying an appropriate set of tools?
I would be very surprised if an “appropriate set of tools” could replicate the effects of a genetic intervention. At least, I would be surprised if you could do so with anything short of AGI or an advanced brain computer interface.
I haven’t spent that much time reading up on non-genetic intelligence interventions (though Gwern has written some pretty good stuff on the topic), but my overall impression is that after you’ve taken care of the basics like having a good diet, exercising, and avoiding lead poisoning you can’t really increase fluid intelligence that much.
Also, any non-genetic interventions that DO work would just stack with the benefits of genetic interventions. So like… why not both?
Curious from a modeling perspective: what improvements would be top of mind for you? Another way to phrase this: if someone else were to try modeling this, what aspects would you look at to tell if it’s an improvement or not?
allow multiple causal variants per clump
more realistic linkage disequilibrium structure
more realistic effect size and allele frequency distributions
it’s not actually clear to me the current ones aren’t realistic, but this could be better informed by data
this might require better datasets
better estimates of SNP heritability and number of causal variants
we just used some estimates which are common in the literature (but there’s a pretty big range of estimates in the literature)
this also might require better datasets
Hey, I found this super interesting and looks like you’ve done your research. I know the good folks over at IndieBio and I think you are at a really good stage to have a chat with them. I think they could be helpful in helping you find a co-founder and initial and follow-on funding for the company. I’m sure there are a number of other synthetic bio accelerators that would be helpful as well, but I have personal experience with them.
That would be very helpful. Would you mind introducing me to them? You can send me an email here: morewronger@gmail.com
This is an interesting post, but it has a very funny framing. Instead of working on enhancing adult intelligence, why don’t you start with:
Showing that many genes can be successfully and accurately edited in a live animal (ideally human). As far as I know, this hasn’t been done before! Only small edits have been demonstrated.
Showing that editing embryos can result in increased intelligence. I don’t believe this has even been done in animals, let alone humans.
Editing the brains of adult humans and expecting intelligence enhancement is like 3-4 impossibilities away from where we are right now. Start with the basic impossibilities and work your way up from there (or, more realistically, give up when you fail at even the basics).
My own guess, by the way, is that editing an adult human’s genes for increased intelligence will not work, because adults cannot be easily changed. If you think they can, I recommend trying the following instead of attacking the brain; they all should be easier because brains are very hard:
Gene editing to make people taller. You’d be an instant billionaire. (I expect this is impossible but you seem to be going by which genes are expressed in adult cells, and a lot of the genes governing stature will be expressed in adult cells.)
Gene editing to enlarge people’s penises. You’ll be swimming in money! Do this first and you can have infinite funding for anything else you want to do.
Gene editing to cure acne. Predisposition to acne is surely genetic.
Gene editing for transitioning (FtM or MtF).
Gene editing to cure male pattern baldness.
[Exercise for the reader: generate 3-5 more examples of this general type, i.e. highly desirable body modifications that involve coveting another human’s reasonably common genetic traits, and for which any proposed gene therapy can be easily verified to work just by looking.]
All of the above are instantly verifiable (on the other hand, “our patients increased 3 IQ points, we swear” is not as easily verifiable). They all also will make you rich, and they should all be easier than editing the brain. Why do rationalists always jump to the brain?
The market has very strong incentives to solve the above, by the way, and they don’t involve taboos about brain modification or IQ. The reason they haven’t been solved via gene editing is that gene editing in adults simply doesn’t work nearly as well as you want it to.
This is more or less our current plan.
This has some separate technical challenges, and is also probably more taboo? The only reason that successfully editing embryos wouldn’t increase intelligence is that the variants being targeted weren’t actually causal for intelligence.
This seems harder, you’d need to somehow unfuse the growth plates.
A nice thing about IQ is that it’s actually really easy to measure. Noisier than measuring height, sure, but not terribly noisy.
More intelligence enables progress on important, difficult problems, such as AI alignment.
It’s hard, yes—I’d even say it’s impossible. But is it harder than the brain? The difference between growth plates and whatever is going on in the brain is that we understand growth plates and we do not understand the brain. You seem to have a prior of “we don’t understand it, therefore it should be possible, since we know of no barrier”. My prior is “we don’t understand it, so nothing will work and it’s totally hopeless”.
Actually, IQ test scores increase by a few points if you test again (called test-retest gains). Additionally, IQ varies substantially based on which IQ test you use. It is gonna be pretty hard to convince people you’ve increased your patients’ IQ by 3 points due to these factors—you’ll need a nice large sample with a proper control group in a double-blind study, and people will still have doubts.
Lol. I mean, you’re not wrong with that precise statement, it just comes across as “the fountain of eternal youth will enable progress on important, difficult diplomatic and geopolitical situations”. Yes, this is true, but maybe see if you can beat botox at skin care before jumping to the fountain of youth. And there may be less fantastical solutions to your diplomatic issues. Also, finding the fountain of youth is likely to backfire and make your diplomatic situation worse. (To explain the metaphor: if you summon a few von Neumanns into existence tomorrow, I expect to die of AI sooner, on average, rather than later.)
This is in fact the plan.
This would be a very big thing in and of itself. Also, it wouldn’t give you much useful information about whether adult editing would work, because most of the uncertainty centers around delivery efficiency, the effect size of edits in adult brains, mosaicism and other things you wouldn’t be able to validate in embryos.
To the best of my knowledge, this is not possible in adults. The growth plates fuse at the end of puberty. This is why bodybuilders taking HGH don’t get taller.
I don’t think gene editing will be able to help with this one. You’d need to swap an X chromosome to a Y or vice-versa. None of the delivery vectors are large enough to fit an entire chromosome. They’re not even close.
And even if you could select a subset of the genes most impactful, you’d have to eliminate the existing chromosome, which is not trivial.
And even if you could do that, it wouldn’t be able to undo male or female puberty.
And even if you could do that I have no idea how this tech could be used to grow different sexual organs, which is what you would ideally want.
MAYBE this could be used to enable endogenous sex hormone production or something. But apart from that, nothing comes to mind for ways this tech could help people who want to transition.
This one could actually be possible. In fact it would probably be easier than intelligence or brain disorders. But your competition is going to be hair transplants and rogaine, which would be difficult to beat on price.
Obviously there are many more examples. That’s one of the exciting things about in-vivo editing. But you have limitations:
The trait must be heritable (true for many things we’d want to change)
We need good genetic predictors for the trait
We would not even attempt the therapy unless the difference was easily measurable.
Yeah, this is what I used to think before I actually worked at a bunch of startups and realized that the efficient market hypothesis doesn’t apply to all markets equally. In reality there are hundred dollar bills lying around everywhere, but most people can only see a few, and some can’t see any.
This is particularly true when there are high barriers to entry, hidden information, many steps of inference to reach a conclusion, and cultural taboos that prevent people from looking. Every one of those is getting in the way here.
Some points:
the 23andme dataset is probably not as useful as you project. They are working from a fixed set of variants, not full genomes or even a complete set of SNPs known to vary. There are certainly many SNPs of interest that just aren’t in their data.
in projecting the gains from discovering further variants that affect intelligence, it’s not clear whether you’ve accounted for the low hanging fruit effect. With these statistical approaches, we obviously discover the variants of largest effect first. Adding millions of additional genomes or genotypes will allow us to resolve thousands of additional common variants, but they are going to be the ones that have really tiny effect sizes.
On the other hand (contradicting point 2 somewhat), quite a substantial fraction of variation in intelligence and other traits is likely due to the genetic load—rare mutations, some likely of substantial effect, all deleterious by definition. Identifying these and their effects is a thorny statistical problem due to their rarity, but if we can, they would actually be very promising edit targets. The advantage being the likely lack of negative side effects, and the fact that the top few for any person would likely be of large effect. Some of them are also probably wide-effect boosts, fixes to fundamental bits of cellular machinery! Downside is that this would be a custom targeting job per person.
The use of ’800 IQ’ is a little grating. The tests only go to 200 or 210 (and are not convincingly normed at that level). Still, fully superhuman, entirely outside the normal human trait range… I guess it’s a fair way to gesture at that.
our predictive models for IQ work significantly better for European or white populations because they were trained on that population. This implies that obtaining a bunch of data for Asian and African populations would allow us to identify additional targets. It surprises me that we don’t have some huge dataset from China, but at least we recently developed a 100K+ genotype of Han individuals, which should turn up some additional hits.
Overall, really promising direction. I appreciate the writeup on new and improved edit methods—I had not been following the field closely, and was unaware we had advanced this much on the previously state of the art CRISPR/Cas9.
It’s possible the source I read was misleading, but last I checked they use SNP arrays with 650k variants, which is roughly all loci with minor allele frequency >1%. That’s enough to make quite a strong predictor, especially since they have a fair number of non-european participants with different linkage disequilibrium (more helpful for pinpointing the causal variant in a cluster).
The simulation accounts for that. That’s why gain per additional edit is logarithmic.
We’ll get better at identifying rare variants with large causal effects soon. UK Biobank just released 500k whole genomes in late November, so we should see the first studies on that data come out in the next few months.
The simulations we ran assume that the dataset only contains variants with minor allele frequencey >1%. Any vairants with lower frequency than that will increase the average marginal effect per edit but aren’t necessary for this tech to work in general.
This is why I specifically used language in the post like “don’t take this too seriously” and “I don’t expect such an IQ to actually result from flipping all IQ-decreasing alleles to their IQ-increasing variants for the same reason I don’t expect to reach the moon by climbing a very tall ladder”
There’s less difference between genetic ancestry groups when it comes to editing than there is for embryo selection. With embryo selection, you can rely on linkage disequilibrium patterns remaining relatively consistent among Europeans to compensate for your uncertainty about which variant in a cluster is causal. You can’t do that with editing.
So getting data from other ancestry groups (particularly Africans, who have the greatest variance in LD structure) will actually editing more efficient for everyone, including Europeans.
The lack of non-European data is slowly being solved, but at the moment I know of no non-European data source that has good IQ phenotype data. There are definitely biobanks and consumer genomics companies who have the data, so they could do it if they want to.
Thanks for the thoughtful comment. I’m glad you enjoyed the post!
I read of a proposal a few months back to achieve brain immortality via introduction of new brain tissue that can be done in a way as to maintain continuity of experience and personality over time. Replenisens , Discussion on a system for doing it in human brains That would perhaps provide a more reliable vector for introduction, as the brain is progressively hybridised with more optimal neural genetic design. Perhaps this could be done more subtly via introduction of ‘perfected’ stem cells and then some way of increasing rate of die off of old cells.
Instead of gene editing could you just construct a ‘perfect’ new chromosome and introduce one or more instances of it into existing neurons via viral injection techniques to increase expression of beneficial factors? No particular reason why we can only have
42🤣46 chromosomes, and this would perhaps side-step difficulties to do with gene editing. Might be a more universal solution too if we could come up with a single or small variety of options for a ‘super’ brain optimising added chromosome.Politically the way to pitch it would be for its life saving/enhancement ability—offered for example to people with low intelligence and educational outcomes to offer them a better chance at happiness in life.
I’d be worried about the loss of memories and previously learned abilities that would come along with “increasing die-off of old cells”.
Also, there isn’t really much extra room in the brain for these new neurons to go. So unless they were somehow a lot smaller I think you’d have to basically replace existing brain tissue with them.
It’s an interesting idea. It seems likely to be substantially more invasive than what I have in mind for the gene editing treatment, but if it actually worked that wouldn’t necessarily be a huge concern.
The thing about large scale interventions like “adding a new chromosome” is that it’s going to be much harder to generalize from existing people what the effects will be.
If we got this technology working REALLY welll, like 99% editing efficiency and no immune issues with redosing, then we could probably try out adding new genes in randomized control trials and then slowly assemble a new chromosome out of those new genes. But I don’t know when or even if we’ll reach that point with this tech.
In the long run digital intelligence will win, and if we miraculously solve alignment and have any agency, we’ll probably just be digital uploads.
Isn’t having extra chromosomes usually bad? https://en.wikipedia.org/wiki/Trisomy
(PS the usual number is 46)
The extra chromosomes have duplicate genes on them. The “excessive number of copies of correct genes” is the hypothesis for why it is bad.
Theoretically a new chromosome with new cellular firmware codes genes that turn off the legacy genes being overwritten. If you had an advanced enough understanding of biology and advanced tools and advanced testing methods you could do this. It would look nothing like today.
I like the premise. I’m glad this is getting researched. But:
Lots of things in the space are understudied and the startup-vibe approach of “we’ll figure this all out on the way because previous papers don’t exist” seems way less likely to work with bio than tech because of the length of iteration cycles. But props if it does?
Black swan effects of polygenic edits
cellular stress if on a large scale?
might be an exception where pleiotropy does actually matter, which would suck. the table in another comment showing correlations between illnesses is pretty convincing however it’s possible there are effects that aren’t quantified there (doesn’t present as diagnosable disease)
???? not sufficiently enmeshed in the bio space but this entire post gives off the vibe of “most of the components are bleeding edge and there aren’t many papers, esp not large scale/long term ones” and I imagine that’ll cause more issues than you expect and streeeetch timescales
given black box bio and difficulty of studying the brain, it’s really hard to tell what’s being left out in studies that measure only change in intelligence/what other things are being affected
We have gotten nowhere near as much as we could out of behavioral interventions (on long timescales) and nootropics, and both of those seem like better areas to put research time into. I don’t actually think a research project of this scale will be faster (for AI safety research etc) than either of those.
counterpoint: this will just make it easier/lower ‘energy’ to apply interventions and is hence worthwhile?? but it’s still so risky that I maintain the above approaches are more worthwhile in the short term
We already expect that too many editor proteins in the cells could be a problem. But that will show up in cell culture experiments and animal studies and we can modify doses, use more efficient editors, and do multiple rounds of editing to address it.
We also know about liver toxicity from too many lipid nanoparticles, but that’s an addressable concern (use fewer nanoparticles, ensure they get into the target organ quickly)
I’m sure there will be others that I don’t expect, but that’s true with literally every new medical treatment. That’s the whole point of running experiments in cell cultures and mice.
We actually have some pretty good studies on plietropy between intelligence and other traits. The only consistently replicated effect I’ve seen which could be deemed negative is a correlation with mild aspbergers-like symptoms.
But you can just look at current people already alive to test the plieotropy hypothesis. Do unusually smart people have any serious problems that normal people don’t?
The answer is pretty clearly “no”. I expect that to continue being the case even if we push intelligence to the extremes of the current human range.
This is true almost by definition for any new technology.
I would be very surprised if a pharmaceutical, or even a bunch of pharmaceuticals could replicate the effects of gene editing. Imagine trying to create a set of compounds that coul replicate the effects of gene editing: you would need thousands of different compounds to individually target the pathways affected by all the variants. And you would need pharmaceuticals that would modulate their activity based on the current state of the cell. After all, that’s how a lot of promoters and repressors and enhancers work; their activity depends on the state of the cell!
I still think people should work on nootropics. And you may of course be right that this won’t be ready before AGI. I’d put the odds at maybe 20%.
But it COULD actually work! And if it did the impact would be absolutely massive! So like… why not? I might as well try.
Torsion dystonia seems to add 10 IQ points. I think there are a few other genetic diseases more common among Ashkenazi Jews that are also associated with higher intelligence.
See here
side comment that I’ve been reminded of: epigenetics *exist(s?)*. I wonder if that could somehow be a more naturally integrate-able approach
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4251063/
I’m sure you’ve already thought of this, and I know nothing about this area of biology, but isn’t it possible that the genes coding for intelligence more accurately code for the developmental trajectory of the brain, and that changing them would not in fact affect an adult brain?
The LessWrong Review runs every year to select the posts that have most stood the test of time. This post is not yet eligible for review, but will be at the end of 2024. The top fifty or so posts are featured prominently on the site throughout the year. Will this post make the top fifty?
Thanks for posting! From a ten thousand foot non-expert perspective I always sort of assumed there wouldn’t be much extra challenge to editing multiple genes at once relative to editing a single gene—if you’re deploying something to go in and make edits to every cell on the body, why can’t it make more than one edit? Or failing that why can’t multiple editing agents be in the same treatment? Interesting to read about the practical challenges and solutions.
Naive question : about immunogenicity, what are the problems with the obvious strategy to counter it ? (target the thymus first to “whitelist” the delivery method).
You know, I actually looked into this at one point.
At the time I didn’t find any obvious reason why it wouldn’t work. But I didn’t spend that much time digging into the details, so my prior is it will be hard for some reason I haven’t discovered yet.
If you could actually find a way to present aribtrary “self-antigens” to T and B cells during the development phase within the thymus, that would be an incredibly powerful technology. It seems plausible to me that we could potentially cure a large percentage of autoimmune conditions with that technology, provided we knew which epitopes were triggering a particular immune response. But I know much less about this area than about gene editing, so it’s entirely plausible I’m wrong.
There’s already a few therapies that basically take this approach; allergy shots are probably the most basic, though I don’t believe they actually do anything with the thymus. The general term for this approach seems to be “Immune Tolerance”.
With a short search, I don’t see anything about reprogramming the thymus.
This is a very fun post, and I wish you all the best. To be briefly negative, it seems more or less completely impossible for this to impact AI outcomes with any reasonable model of the world at all, because it will not provide enough enhancement quickly enough. But it’s a cool direction, and worth pursuing as its own science!
If we get to AGI in the next 5 years I think you’re right. 5-10 years and I’d give it maybe a 20% chance of having a large impact. 15 years maybe a 40% chance. And if we have 20 years or more then iterated meiotic selection may have time to work.
Have people thought about doing gene editing stuff at proposera? Seems legal there?
Regulatory issues are not the main bottleneck at the moment. It’s possible we’d consider doing trials down there in a few years when we are ready for actual human testing. But there’s still a substantial amount of building and validation to do before we need to think about that.
This seems great. Better to have superintelligent people than superintelligent AI.
I only have one concern, which is how the IQ is measured? I was assessed as a child and told I was average IQ and that I’d probably never learn English or Mathematics. But when I was 12 I scored 125 on the Mensa.dk test, and a while ago I scored 150+ on BRGHT so I’m not as dumb as the system believes. Not that I would recommend making any more of me.
That type of IQ tests only measure one type of intelligence, and I believe that verbal intelligence is more important for regular functioning, and that memory and working memory shouldn’t be neglected either (they are part of what made Neumann such a monster). I will even claim that most teachers won’t be able to tell the difference between a student with great memory and a student with great spatial intelligence until at least college.
These are all just grouped under “IQ”, and, I would argue, often measured quite poorly. Not to mention that the concept of IQ is under a lot of political pressure and emotionally/morally motivated criticism from people who barely understand the subject, like that video from Oxford where the speaker seems to think that fluid intelligence is the same thing as creativity.
I also hope that not all popularized IQ scores are included in that dataset. There are some false positives, like Christopher Langan (I don’t know how to format links): https://www.reddit.com/r/slatestarcodex/comments/a38tik/the_mega_test_and_bullshitchristopher_langan/
But the genes of previous geniuses who actually contributed to humanity, are valuable
What’s your best guess for what percentage of cells (in the brain) receive edits?
Are edits somehow targeted at brain cells in particular or do they run throughout the body?
From my conversations with people working on delivery vector, I’d guess you can probably get 20-40% with current AAVs and probably less with current lipid nanoparticles.
But that percentage has been increasing over time, and I suspect it will continue to do so. That’s why I estimated that brain cells would, on average, receive 50% of the edits we attempt to make; by the time we’re ready to do animal trials that is probably roughly where we’ll be at.
This is addressed in the post, but I understand that many people may not have read the whole thing because it’s so long.
The set of cells that will be targeted depends on the delivery vector and on how it is customized. You can add custom peptides to both AAVs and lipid nanoparticles which will result in their uptake by a subset of tissues in the body.
Most of the ones I have looked at are taken up by several tissues among which is the brain. This is probably fine, but as stated in the appendix there’s a chance expression of Cas9 proteins in non-target tissues will trigger the adaptive immune system.
If that did turn out to be a big issue, there are potential solutions which I only briefly touched on in the post. One is to just give someone an immunosuppressant for a few days while the editor proteins are floating around in the body. Another is to selectively express the editors in a specific tissue as specified by the mRNA transcribed uniquely in that cell type.
The latter would be a general purpose solution to avoiding any edits in any tissues except the target type, but would reduce efficiency. So not something that would be desirable unless it’s necessary.
@TsviBT
Quite confused about the non-coding region edit hypothesis.
Either you mean “non-coding” as in “regulatory” in which case… wouldn’t off-target mutation be just as bad?
Or do you mean “non-coding” as in “areas with an undetermined role that we currently assume are likely vestigial”—in which case, wouldn’t the therapy have no effect since the regions aren’t causal to anything, just correlated? [Or, in the case where I’d have an effect, we ought to assume that those “non-coding” regions are quite causal for many things and thus just as dangerous to edit]
Non-coding means any sequence that doesn’t directly code for proteins. So regulatory stuff would count as non-coding. There tend to be errors (e.g. indels) at the edit site with some low frequency, so the reason we’re more optimistic about editing non-coding stuff than coding stuff is that we don’t need to worry about frameshift mutations or nonsense mutations which knock-out the gene where they occur. The hope is that an error at the edit site would have a much smaller effect, since the variant we’re editing had a very small effect in the first place (and even if the variant is embedded in e.g. a sensitive binding site sequence, maybe the gene’s functionality can survive losing a binding site, so at least it isn’t catastrophic for the cell). I’m feeling more pessimistic about this than I was previously.
I don’t particularly see why the same class of errors in regulatory regions couldn’t cause a protein to stop being expressed entirely or accidentally up/down-regulate expression by quite a lot, having similar side effects. But it’s getting into the practical details of gene editing implementation so no idea.
A bad enough error in a regulatory region could cause a protein to stop being expressed. But the insertion or deletion of a single base pair is not nearly as devastating.
Let me explain by talking through how a promoter works.
Promoters sit upstream of a region of the genome that codes for a protein. They generally serve as a binding site for a very important enzyme called RNA polymerase, whose job it is to transcribe DNA into mRNA, which can then be exported from the nucleus and turned into proteins.
You can delete a letter from a promoter and RNA polymerase will still be able to bind. The “binding affinity” (meaning the strength of the bond) will be affected by this deletion, but except in rare circumstances it will still work.
You can see this reflected in the distribution of insertion and deletions throughout the genome; there’s not that many in coding regions, but there are tons in non-coding regions (on the order of 3-5 million).
Oh, ok, the mechanism is familiar to me and in hindsight this makes sense !
But then, my follow-up would be, if all you are doing is up/down-regulating certain proteins or regions encoding several proteins wouldn’t you be able to more easily either get the proteins or plasmids or RNAviruses expressing the proteins into the brain ? Which would be temporary but could be long lasting (and cheap) and would not pose this risk
great article. I hope you realize your startup research/idea. One comment, I think the salaries derail the whole budget plan, afaik from startup world I have been involved, founders make big sacrifices to get their thing going in return for a big equity in the startup they believe someday will become a unicorn.
This is super interesting and I have a question:
How difficult would it be to also apply this to the gamates and thus make any potential offspring also have the same enhanced intelligence (but this time it would go into the gene pool instead of just staying in the brain)? Does the scientific establishment think this is ethical? (Also, if you do something like this, you reduce the homogeneity of the gene pool which could make the modified babies very susceptible to some sort of disease. Would it be worth it to give the GMO babies a random subset of the changes to increase variation?)
Not very difficult. In fact it would be easier with fewer things that could go wrong, and a much greater ability to validate.
It depends on who you ask, but the general answer is “no”, for reasons no one can ever articulate very clearly. In general most scientists are just extremely risk averse to doing anything that involves reproduction. See the section in the post titled “Vague associations with eugenics make some academics shy away”
Also, if you do something like this, you reduce the homogeneity of the gene pool which could make the modified babies very susceptible to some sort of disease.
Brain editing wouldn’t really affect the immune system.
Frankly of all the potential things we could edit, the one I am most hesitant about is the immune system. There are unique concerns with immunity that don’t apply to other areas: for example, we know there are genetic variants whose allele frequencies increased substantially during the black plague because they conferred significant resistance to the disease.
Some of those variants increas the risk of autoimmune disorders as well!
And given the infrequency of huge pandemics, you can’t do as good of a job making tradeoffs between decreased autoimmune risk and pandemic susceptibility.
There is a HUGE amount of variance in human immune systems. There’s so much variance in the Major Histocompatibility Complex that SNP arrays have noticeably increased error rates when we try to genotype people.
IMO no, but you actually bring up an interesting point: could we generate more genetic diversity by giving huge numbers of babies random variants and seeing what they do?
I think you probably could but it would take a very, very long time to suss out the full effect and incorporate it into your predictors for the next generation. Unless we have a global ban on AI development for many decades, I don’t see any point.
As far as giving different people a different subset of trait-affecting alleles, sure. You could do that in embryos.
With adults though, the bottlenecks due to editing efficiency, no effects from genes that affect development and others mean that you probably don’t want to “deselect” that many variants.
This makes it sound like CAR-T is gene editing, but it isn’t. Instead of editing a gene, it introduces a new one (a chimeric T-cell receptor). Although some companies are working on gene editing to enhance CAR-Ts.
I was under the impression that the new gene usually integrated into the cell’s genome. But that impression was from a conversation with GPT-4 so perhaps I’m mistaken. Or perhaps new gene insertions are not considered gene editing?
It does integrate into the genome. It’s gene therapy, but not gene editing (which means editing an existing gene).
What about this? https://www.nature.com/articles/s41598-023-39777-0
Would these edits result in demethylated DNA? A reversion of the epigenome could allow expression of infant genes. There may also be robust epigenomic therapies developed by the time this project would be scalable.
Just as you mentioned that academics’ aversion from this area, I think genomics companies would be reluctant at best to ask their customers for test scores. Perhaps it wouldn’t be bad PR once the public is more concerned about existential AI. Governments might be more willing to provide data.
I see the biggest problem not on the technical side of things, but on the social side. The existing power balance withing the population and the fact that it discourages cooperation is in my opinion a much bigger obstacle to alignment. Heck, it prevents alignment between human groups, let alone between humans and the future AGI. I don’t see how increased intelligence of a small select group of humans can solve this problem. Well, maybe I am just not smart enough.
Do you think there’s an Algernon tradeoff for genetic intelligence augmentation?
Probably not? The effect sizes of the variants in question are tiny, which is probably why their intelligence-promoting alleles aren’t already at fixation.
There probably are loads of large effect size variants which affect intelligence, but they’re almost all at fixation for the intelligence-promoting allele due to strong negative selection. (One example of a rare intelligence promoting mutation is CORD7, which also causes blindness).
I took the pfizer vaccine and I found the section explaining how the mRNA vaccine works both illuminating and disturbing. Getting cells to generate and display the spike element of the virus, causing inflammation—and potentially cell death is perhaps all fine in the muscle tissue of the shoulder, but what happens if the mRNA travels further to different tissue like the heart? Upon reading how the vaccine actually works I have become a lot more suspicious of potential bad effects of the vaccine—previously I was under the naive impression that the vaccine is more like putting some inactive element on the body for the immune system to recognize.
The analogy of mRNA behaving like root access to cells using their machinery to create proteins—is powerful . I am now a lot more sceptical of the vaccine work, because now I have a better understanding of just how innovative crazy the science is.
Man, if you think the vaccine is scary just wait until you hear about what COVID does.
In all seriousness, you shouldn’t find mRNA vaccines any scarier than the J&J vaccine or the Novavax vaccine. J&J puts DNA for the spike protein in a modified adenovirus, which then enters your muscle cells, breaks out into the cytoplasm, and injects the DNA payload into the nucleus. Then RNA transcriptase makes mRNA out of that DNA, which is exported from the nucleus into the cytoplasm where it is turned into the spike protein.
Novavax just skips the entire manufacturing process; they just inject the spike protein directly into your body.
I’m not an expert on COVID vaccines, but from everything I have read, the rare dangerous side-effects like myocarditis seem to come from the spike protein itself triggering a dangerous immune response in some very small percentage of people.
But like… you can’t avoid that with any vaccine. And the incident of getting myocarditis or a worse condition from COVID itself (especially if you’re unvaccinated) are way, way higher than getting it from the vaccine.
Vaccines are mostly just a cost-benefit analysis; if your odds of damage are higher from the virus (adjusting for the probability of infection) compared to the vaccine, you’re better off getting vaccinated. That will be the case for virtually everyone. Maybe there’s some case for young children to not get the vaccine because the virus itself is so much less dangerous for them. But that’s about it.
I mean this is all kind of a moot point now because everyone has either been infected or vaccinated already, but if your main takeaway from this post was that mRNA vaccines are dangerous I would be pretty disappointed.
Qualifications to Consider
These are merely my qualifications I would place on this seemingly very interesting and highly well informed, well researched and competent idea the author has raised. Overall, these qualifications are of the “meta” sociological and political intersections onto the trait being enhanced here. Before presenting them, I would also like to state that the novel characteristics and approach to this subject that the author, who is not formally educated in biology and free of its academic milieu, is suggestive that the culture that surrounds subjects like this functions as a smothering dogma holding back innovation not advancing it as many academics would prefer to tell themselves.
Qualifications:
1. The use of biotechnology to enhance intelligence raises ethical questions regarding fairness and equality. It would create an uneven playing field in terms of astronomical cost or not be FDA approved, as only those who can afford the enhancement may have access to it, exacerbating existing social inequalities like jet fuel on a dumpster fire.
2. Modifying genes to enhance intelligence could have unforeseen side effects or unintended consequences. Genetic modifications can have complex and unpredictable effects on various aspects of an individual’s physical and mental health, potentially leading to unforeseen negative outcomes.
3. Intellectual diversity is valuable for society as a whole. If everyone were genetically enhanced to have significantly higher intelligence, it may lead to a loss of diverse perspectives, creative thinking, and alternative problem-solving approaches that contribute to the richness of human society.
4. Individuals who choose not to undergo genetic enhancement for intelligence may face stigmatization or discrimination in a society that places a high value on intelligence. This could create a divide between enhanced and non-enhanced individuals, leading to social tensions and inequality.
5. Focusing solely on increasing intelligence may lead to an imbalance in human development. Other important qualities such as emotional intelligence, creativity, empathy, and social skills may be undervalued or neglected, potentially resulting in a society that lacks holistic development. As we can deduce from the late 19th and early 20th centuries, pure application of intelligence even with the urging of now socially disregarded religious mores to empathy for others, there tends to be some really horrific wars that result and that creates incentives for weapons systems to be developed that could lead to the extinction of the species. As has been said by a wise sage of the Levant long ago, “man cannot live by bread alone.”
6. The long-term effects of genetically enhancing intelligence are currently unknown. It would require extensive research and testing to fully understand the potential consequences, including any negative impacts on individuals and the broader population. This would also be the means the gatekeepers are most likely to hold these things back using, if first they don’t use the patent court racket first (see below for thoughts on that specifically intended for the author’s consideration).
7. Introducing widespread genetic enhancement for intelligence could have far-reaching societal implications that are difficult to predict. It may disrupt social dynamics, undermine traditional educational systems, and challenge existing notions of intelligence, potentially leading to unintended societal consequences. This being the primary fear of the plutocratic elite, who already game the economy, government and society itself (via mass media and implanted insecurities picked up from them as hypnotic triggers and post-hypnotic suggestions among other marketing tactics) to stabilize their ranking and prevent too much movement in or especially out of their ranks and widespread social change, even positive for the vast majority, would likely amount to shake up in their ranks of the order their individual best interest would make them inclined to reject.
8. Assuming intelligence is primarily IQ, which is how quickly and effectively an individual comes up with a solution for a novel problem, does not fully encapsulate the notion meant by the word intelligence. The exact conceptual boundaries are open to extensive debate on this subject in particular and are charged by the raging insecurities of those attempting its definition that have rendered this concept outside of post-modernity’s ability to adequately define with any precision. Raising problem solving capacity without subsequent and proportional increases in the other elements that partially factor into intelligence may actually lead to a reduced overall intelligence in the treated population, or the increase of problem solving capacity might have a long term effect of reducing the other elements such that this primes a (steeper) downward trend in the intelligence of subsequent generations.
The Patent Court Racket
You are clearly very intelligent and apparently learn quickly, so now is the time to learn how to write patent applications for any and all processes you think evidently implied by these ideas or face paying a licensing fee to conduct research along lines you came up, if the patent holder even allows that much and doesn’t shovel the idea into the filing cabinet of doom. Patents are granted for technology and processes employing them that are evidently not possible, or if they are not exploited, otherwise patents for controlling peoples’ emotions using flickering of CRT monitors should probably be of a greater concern to the average person. Your intelligence being of a “raw” quality and your idea coming off with little self-righteousness also implies that you can be bought, so getting the patents might turn out to be rather lucrative because this sort of thing once the patent application is published will generate some profound interest for reasons discussed above. In the future, patent first and make announcements second, if for no other reason than
This is of course something we’ve thought about. It’s a little hard to think too seriously about this so long as we stay on track to develop AGI, which will the gap between people due to genetic differences seem very small by comparison.
But if I ignore that for a moment, I think the best way to tackle this is to make sure the technology is to ensure the per unit cost is not too high (ideally < $10k), and to perhaps offer some innovative payment plans such as taking a percentage of people’s future earnings over the level they currently make for some period of time. So for example, instead of paying for the treatment directly, you might agree that for any money you make in excess of your current income, you pay the company 30% of it for 5 years.
This is of course somewhat of a concern, but most of the research I’ve read shows that whatever plieotropy exists between intelligence and other triats mostly works in your favor. In other words, the genes that increase intelligence generally tend to slightly decrease disease risk, violent behavior, and other traits generally considered negative.
The only exception to this I’ve seen this far is mild aspergers, the risk of which seems to be slightly increased by the same genes that affect intelligence. To the extent that this is a problem, we could simply identify the subset of genes that increase intelligence without increasing aspergers, or in addition to editing genes to increase intelligence, also edit genes to keep the risk of aspbergers constant at the same time.
There’s 8 billion people on the planet. It’s going to take a very long time to edit even a million people. So I don’t think this should be a concern for at least another 50 years, by which point the question will probably be moot unless we have a global pause on AI development.
Yes. I don’t really have a society-wide answer to this yet other than the good old “treat other people well”. This is already an issue with just natural variation in abilities, though gene editing would undoubtedly exacerbate it.
We don’t actually plan to solely focus on intelligence. In fact the first targets will be polygenic brain diseases like Alzheimers or treatment resistant depression. I also think it would be good if we could modify other traits such as conscientiousness, tolerance to sleep deprivation and others.
We have very smart people around right now. They seem to be doing fine. Maybe there are “long term consequences” to modifying people to be outside the human range, but we probably won’t push that far outside the limits of what naturally occurs.
It’s also plausible that we could reverse some of the effects of editing with another round of edits designed to push in the opposite direction.
I agree of course. The post was already over 30 pages long, so I decided not to discuss other forms of intelligence. But in reality those would be of interest as well.
However, the hard part with considering any of that is that we don’t currently have the phenotype data to create predictors of JUST reading ability or JUST math ability.
I spoke with a few biologists before publishing this post, each of which informed me that I don’t have any truly novel ideas here. I have put together several existing ideas in a novel way, but it’s doubtful they are patentable.
We WILL file patents after we start doing lab work.
Hi,
Since I have read your entire post on intelligence boosting I had to DM you.
I ask you as a fellow human being who errs just as much as everybody else to read this with open and calm mind, refraining from immediate judgement. I have tried to be as critical as possible not to bully you, but in order to point out some holes in this post which I hope in the end will do nothing but improve it.
It seems to me that there are some very significant obstacles to this. You have addressed 2 of them, however solutions you provided are not nearly solutions. I think you are a bit too optimistic about this, although I like optimism, we can’t allow ourselves to chase a fictional dream just because we are slaves to our physiology of dopamine drive which works on the basis of least resistance path. So we are always inclined to favor a bit undefined but promising solutions over real, non-ambiguous data which clearly demonstrates the solution will not work.
I dare say this because I have read too much statements in this post like: with further research this can be solved or One has to believe. (Believe is what hopeless do)
I understand you are deeply invested in this, and I really wish you were right, but there are just WAY TOO MANY UNSOLVED PROBLEMS here and WAY TOO LITTLE DATA on how the body and especially each cell works in the brain—not all cells work the same, yet every single one of them is essential. It is like we have no map of the environment but wish to visit a castle of king Arthur.
I don’t think people are trying to pick the low hanging fruit with monogenic diseases, these diseases are simply a priority for us to solve, coupled with sufficiently advanced technology and knowledge to actually solve them. However delivering 100 edits per single lipid particle is like saying we are able to design, build and actually deliver a fully functional warp 7 capable spaceship in 2 weeks, give or take 3 days with current knowledge.
I truly think we should talk about this and I agree with you on many of your points, but before solving a biological limit to intelligence perhaps we should first try to reach it. Imagine if our education systems worked efficiently enough to push average IQ to 160, wouldn’t it be interesting. And besides, what makes you think genetics will magically triple our IQ, if we are not capable of reaching IQ of 150 as a society than how on Earth do you think we will be able to reach 800. Like you are trying to build a car with max speed of 290 mph, while you don’t even have a driving license let alone know how to control a beast capable of 150 mph.
KEY POINT: there is no magic solution, if it is too good to be true, it usually is untrue.
Good luck and thank you!